

��������
��ϵ��ʽ
�����ݺᣨ���ڣ���������˾
ȫ���������ߣ�4006-010-725
����:��ɽ�������ֵ�����·������·������֮����
�ͷ�QQ��574472821
��:13077829315
�� ��:130-7782-9315
�� ��:shbsfw#126.com
��ַ��www.gdhdgw.com
���Ŷ�̬
������ʳƷ����ѧ��������淶����������壩��ȫ��
����ʱ�䣺2019-6-14 ¼�룺���ڻ������� ��Դ�����ڻ�������

Ŀ ¼
��һ����
General Principles
һ���������ݺ����÷�Χ
Content and Scope of Application
1.���淶�涨�˱���ʳƷ��ԭ�ϵ�����ѧ����Ҫ��ļ�����Ŀ��������
2.���淶�����ڱ���ʳƷ��ע�ᡢ���˺ͱ������顢�ල���顢���ռ�⼰���������Ŀ��ȷ���ͷ�����ѡ��
��������Ҫ��
Basic Requirements
1. ������ʳƷ��Ӧ����GB 16740��ʳƷ��ȫ���ұ� ����ʳƷ��������Ҫ����1���м����Ŀ�Ƕ��ñ��IJ���涨��
2. ����ʳƷ��ʳƷ���Ӽ���ʹ��Ӧ����GB 2760�Ĺ涨�ͣ����йع涨���������ɸ���ʵ����Ҫ������Ʒ�䷽���ϳ�ɫ�ء�����������ζ�������������ĺ�����
3. ����ʳƷ�İ�װ����Ӧ����GB 4806.1��ʳƷ��ȫ���ұ� ʳƷ�Ӵ����ϼ���Ʒͨ�ð�ȫҪ����Ҫ����
4. ���з�������IJ�Ʒ�������Դ�����ռ����Ⱦ�Ӧ������Ӧ�Ĺ��ұ�����ҵ�����й�Ҫ��
5. ���������������ʳƷ���ʱ���걨��λӦ�ṩ�ò�Ʒ���䷽����������Ʒ����Ҫ��Ч�ɷ�/��־�Գɷּ�ⷽ���о���֤�����������
6. ���뵥λ�ڶԲ�Ʒ���й�Ч�ɷ�/��־�Գɷּ��ʱ������ұ������ṩ�ļ�ⷽ���������ڸò�Ʒ����ʱ��������ѡ�淶�ڶ������ṩ�ļ�ⷽ���������б�Ҫ�ķ���ѧ�������о�����淶�����ṩ�ļ�ⷽ���������ڸ���Ʒ����ʱ��������λ��ѡ����ҵ����������Ȩ�������������вⶨ����������Ҫ�������������о���ע�����������������Ͻ�����ˣ���Ҫʱ�����й���֤�ͷ���ȷ�ϣ����걨��λ�ṩ�ķ������ʺ��ͼ����Ʒʱ��ע�����������������ģ�Ӧ���й���������걨��λ����������о����ṩ�������ٶ��ͼ���Ʒ�������飬ȷ�����鷽�����ͼ��Ʒ����Ҫ���й涨�ķ���һ�¡����˼������Ӧ�����걨��λ�ύ�ļ��鷽�����м��鲢���߸��˼��鱨�档
7. ����ʳƷ��ԭ�Ϻ���Ӧ���ϡ�����ʳƷע���뱸�������취����ԭ����������Ҫ��Ĺ涨����������Ӧ��ʳƷ���ұ���ԭ����Ϊֲ����ȡ�����ԭ�ϼ����ϼӹ�������ʹ�á�������������ܼ�ʱ���漰���л��ܼ�Ӧ����GB 2760��ʳƷ��ȫ���ұ� ʳƷ���Ӽ�ʹ�ñ�����¼C��ʳƷ��ҵ�üӹ�����ʹ�������涨����������ر���Ҫ������ҵ�ɸ��ݲ�Ʒ����������Ҫ�����ñ��淶�е������ֱ���ʳƷ��ʮһ���ܼ������IJⶨ�������ܼ������������ԭ�ϼ���Ʒ�ļ���Ҫ��
8. ��Ը��Ч�ı���ʳƷ������ձ��淶���IJ����˷ܼ���Υ���ɷֲⶨ�������зǷ�����ɸ�顣ͬʱ��������䷽����������ȷ����ԭ�ϴ��������ָ�꣬Ӧ��������Ŀ�������������������Ϊ�Ƿ����ӽ�����ͣ�������쳣�⡣
9. ��ͬ���ͼ��ض�ԭ������Ʒ������ѧ��������Ӧ�������淶���岿���й涨����Ӧ�����Ŀ��
10. ����ʳƷӦ�������Ʒ�䷽���걨�ı�����������Ӧ�Ĺ�Ч�ɷֻ���־���ɷ֣��걨ʱ���ṩ�䷽����Ҫԭ�������Ĺ�Ч�ɷֻ���־���ɷ��ļ�������췽��������ѧ��֤��������������г���ԭ�����ͼ���Ӧ�����Ŀ��������ԭ��Ϊ���IJ�Ʒ������й涨����Ŀ��
11. ��ͨʳƷ��̬��ƷӦ��Ⲣ�ƶ���������������ƫ��ָ�꣬��Ҫ���ղ�Ʒ�������涨������С��װȷ��������ݣ�ָ��Ӧ���ϡ�������װ��Ʒ�����������������JJF 1070���涨�����й�ҩ�䡷“�Ƽ�ͨ��”��������ӦҪ��IJ�Ʒ���ͣ�Ӧ��Ⲣ�ƶ�װ���������������ָ�ָ꣬��Ӧ���Ϲ涨��װ������ֻ��������������������ϣ��罺�ҿǵȡ�
12. ��С���õ�Ԫ���ж��Ը���������IJ�Ʒ���罺�ң��书Ч�ɷֻ���ָ��ɷ֡�ũҩ������ˮ�֡��ҷֵ�ָ����ȥ��������ϣ����ҿǣ���������Ϊ��ⵥԪ�����ڷǷ�����ҩ��ؽ���������ɫ�أ�����ϴ���ɫ��������Ҫ���������⣬���������ȷ��ʶ��ؼ�ⲿλ��
13. ���涨���ü������Ŀ������������������ϼ���������վ�����ֵ���ͽ����������������¼������ע��“δ����������ֵ”��������������ϡ����������¼������ע��“�����С�ڶ����ޣ�������ֵ�������ֵ”��
14. ע���������Ӧ���չ�����ع涨�ͱ���Ҫ������Ʒ��������������ؽ����ȶ���������ƺ��о���ͨ���ȶ������飬������Ʒ�ڲ�ͬ���������£����¶ȡ����ʪ�ȵȣ��ĸй١���ѧ������������ѧ������ʱ��������仯�̶Ⱥ��ɣ��Ӷ��ж���Ʒ��װ�����������ͱ������ڵ��ȶ��ԡ���Ʒ�ȶ����ص㿼��ָ�꣬��Ҫ�������𡢸й١��������ʱ�ޣ���ɢʱ�����ܻ����ȣ���ˮ�֡�pHֵ����ۡ�������ֵ��������ء���������ָ���е���Ч�ɷ�/��־�Գɷ����洢������������ʱ���������仯��ָ�ꡣ��Ʒ���ȶ����ص㿼��ָ�꣬��Ҫ�����ҷ֡���Ⱦ���Ǧ�����顢�ܹ��ȣ���ũ�У������������ε���ȣ���������ر������й涨���������Ƶĺϳ�ɫ�غ���ζ�����洢������������ʱ�䲻�����仯��ָ�꣬�Լ�������ر������й涨���������ƵĿ�������ָ�ꡣ
�ڶ�����
���鷽��
and Iconic Components
һ������ʳƷ�к쾰���յIJⶨ
Determination of salidroside in health food
1 ��Χ
�������涨���Ժ쾰��Ϊ��Ҫԭ�ϵı���ʳƷ�к쾰���յ�Һ��ɫ�ײⶨ������
�����������ڱ���ƷʳƷ�к쾰���յIJⶨ��Ҳ�����ڱ���ʳƷ���Ҵ��IJⶨ��
2 ԭ��
�������״�������ȡ����0.01 mol/L �����-�״�Ϊ�����ࣨ80+20�������ø�ЧҺ��ɫ��������������⣬���ݱ���ʱ�䶨�ԣ������������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1 �����( CH3COONH4 )��
3.1.2 �״�(CH3OH)��ɫ�״���
3.1.3 �״�(CH3OH)��
3.2 �Լ�����
�������Һ (0.01 mol/L)����ȡ0.77 g���������������ˮ�ܽ⣬��ˮ������1000 mL����0.45 μmˮ������Ĥ���˺��á�
3.3 ��Ʒ
�쾰����(C14H20O7)������≥98%��
�Ҵ���C8H10O2)������≥98%��
3.4 ����Һ������
3.4.1 �쾰����������Һ(2.0 mg/mL)�� ȷ��ȡ�쾰���ձ�Ʒ0.02 g�� 10 mL ����ƿ�У��ü״���3.1.2���ܽⲢ�������̶� ��ҡ�ȡ�
3.4.2 �쾰����������Һ�����쾰����������Һ��3.4.1���ü״���3.1.2��ϡ���Ʊ�һϵ�б���Һ����ϵ��Ũ��Ϊ0mg/mL��0.01 mg/mL��0.02 mg/mL��0.05mg/mL��0.20 mg/mL��0.50 mg/mL������ʱ���ơ�
3.4.3 �Ҵ�������Һ(2.0 mg/mL)��ȷ��ȡ�Ҵ���Ʒ0.02 g�� 10 mL ����ƿ�У��ü״���3.1.2���ܽⲢ�������̶� ��ҡ����
3.4.4 ϵͳ��������Һ��ȷ��ȡ�쾰���ձ�����Һ��3.4.1�����Ҵ�������Һ��3.4.3����0.5mL�� 10 mL ����ƿ�У��ü״���3.1.2��ϡ�����̶� ��ҡ����
4 �����豸
4.1 ��ЧҺ��ɫ���ǣ� ��������������UV����
4.2 ��������ϴ����
4.3 ������ƽ������Ϊ0.01 mg��0.1 mg��
5 ��������
5.1 �����Ʊ�
5.1.1 ������Ʒ��ȡ20������Ƭ�������������з��顢���ȣ�ȷ��ȡ����������Լ���쾰����2.5mg���� 25 mL ����ƿ�У�����״���3.1.3��Լ20mL��������ȡ30 min�����������£��ü״���3.1.3���������̶ȡ����Ⱥ�0.45 μm��Ĥ���ˣ���Һ��ɫ�����á�
5.1.2 Һ����Ʒ��ȷ��ȡ����ҡ�Ⱥ��������Լ���쾰����2.5mg����25 mL����ƿ�����ü״���3.1.3���������̶ȡ����Ⱥ�0.45 μm��Ĥ���ˣ���Һ��ɫ�����á�
5.2 ɫ�ײο�����
5.2.1 ɫ������C18��������250mm���ھ�4.6mm����������5 μm����ͬ������ɫ������
5.2.2 �����ࣺ�������Һ��0.01 mol/L��-�״���80+20����
5.2.3 ���٣�1.0 mL/min��
5.2.4 ���£�25 ����
5.2.5 ��Ⲩ����215 nm��
5.2.6 ��������10 mL��
5.2.7 ϵͳ������������ȡϵͳ��������Һ��3.4.4��10 mL��ע��Һ��ɫ���ǣ���¼ɫ��ͼ���쾰���շ����Ҵ���ķ����Ӧ����1.5��
5.3 �����ߵ�����
����ϵ�й���Һ��3.4.2���ֱ�ע���ЧҺ��ɫ�����У��ⶨ��Ӧ��ɫ����������Ա�����Һ��Ũ��Ϊ�����꣬�Է��������Ϊ�����꣬���Ʊ����ߣ�����ҺҺ��ɫ��ͼ����¼A��ͼA.1����
5.4 ������Һ�IJⶨ
����������Һ(5.1.1��5.1.2)ע��Һ��ɫ�����У��Ա���ʱ�䶨�ԣ���÷�������ߣ����ݱ����ߵõ�����Һ�쾰���յ�Ũ��(��Ʒ��ҺҺ��ɫ��ͼ����¼A��ͼA.2)��
6 ��������ı���
�����к쾰���պ�����ʽ(1)����:
C ×V
X = × 100 .......................................(1)
m
ʽ�У�
X �������к쾰���յĺ�������λΪ����ÿ���˻����ÿ��������mg/100g��mg/100mL����
C—�ɱ����߲�ô�����Һ�к쾰���յ�Ũ�ȣ���λΪ����ÿ������mg/mL����
V—��Ʒ�Ķ����������λΪ������mL����
m—��Ʒ������λΪ�˻������g��mL����
100—��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
8 ����
��������Ϊ 1 g���������Ϊ 25 mLʱ���쾰���յļ����Ϊ50m g/g��
��¼ A
����Һ��������Һ����Һ��ɫ��ͼ
A.1 �쾰���պ��Ҵ�����Һɫ��ͼ����ͼ A.1��
ͼ A.1 �쾰���պ��Ҵ�����Һ��0.1mg/mL��ɫ��ͼ
A.2 ���к쾰���պ��Ҵ���������Һɫ��ͼ����ͼ A.2��
ͼ A.2 ���к쾰���պ��Ҵ���������Һɫ��ͼ
��������ʳƷ�д����صIJⶨ
Determination of allicin in health food
1 ��Χ
�������涨�˱���ʳƷ�д����صIJⶨ������
�������������Դ��⼰��ӹ�ƷΪ��Ҫԭ���Ƴɵı���ʳƷ�д����صIJⶨ��
2 ԭ��
�������л��ܼ���ȡ��ͨ������ɫ����⣬�Ա���ʱ�����ԣ���귨������
3 �Լ�������
ע����������˵���������������Լ���Ϊ��������
3.1 �Լ�
3.1.1 ��ˮ�Ҵ�(C2H5OH)��
3.1.2 ������(CH3(CH2)4CH3)��
3.1.3 ��ˮ������(Na2SO4)��
3.2 ��Ʒ
������(C6H10S3)������≥99.0%��
3.3 ����Һ����
3.3.1������������Һ(10.0mg/mL)����ȡ100.0mg�����ر�������10mL ����ƿ�У��������鶨�����̶ȣ�ҡ��������Һ���ڱ����б������졣
3.3.2������������Һ(1.0mg/mL)����ȡ������������Һ 1.0mL�� 10mL����ƿ�У��������鶨�����̶���ҡ�ȣ���Ũ��Ϊ1.0mg/mL�ı�����Һ������ʱ���ơ�
4 ����
4.1 ����ɫ���ǣ�������������ӻ������( FID)��
4.2 ��ƽ������Ϊ 0.1 mg �� 0.01 mg��
4.3 ������ϴ����
5 ��������
5.1 �����Ʊ�
5.1.1 ��������
��ȡ�������Ͼ��ȵ����������������������������Լ5mg����ȷ�� 0.0001 g����5mL����ƿ�У�����ˮ�Ҵ�2.5mL������������20min��ȡ����ȴ���������������鶨�ݣ�ҡ������0.45μm����Ĥ���ˣ����ϻ���������
5.1.2 ��״����
��ȡ�ѻ�Ͼ��ȵ���״�����������������������Լ5mg����ȷ�� 0.0001 g����5mL����ƿ�У����������ܽⲢ���ݣ�ҡ�������ϻ������á�
5.1.3 ��ˮҺ������
������ȡ����Ͼ��ȵĴ����������������������Լ10mg�������ڷ�Һ©���У���5mL��������ҡ��ȡ1min�����÷ֲ㣬ȡ�ϲ���Һ����ˮ�����ƣ���ȡ��������������������ϴ��ˮ�����ƣ��ϲ���ͬһ10mL����ƿ�У������鶨����ҡ�������ϻ������á�
5.2 ɫ�ײο�����
5.2.1 ɫ��������5%-������-���۹�����̶���������30m���ھ�0.25mm��Ĥ��0.25μm������ͬ������ɫ������
5.2.2�������¶��� ��ʼ�¶�100������3���ӣ� 10��/min�ٶ�����150��������20��/min�ٶ�����200��������20min��
5.2.3 �������¶ȣ�220�棻������1 μL��
5.2.4 ������¶ȣ�250����
5.2.5 �������ߴ�����������1.0mL/min��
5.2.6 ������40mL/min ��������400mL/min��
5.3 �ⶨ
�� 1 μL ������Һ(3.3.2)����������Һ(5. 1)ע������ɫ�����У��Ա���ʱ�䶨�ԣ���÷�������ߣ������������߷�������(����Һͼ������ͼ����¼A)
6 ��������ı���
�����д����غ�����ʽ(1)����
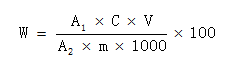 ..........................................(1)
..........................................(1)ʽ�У�
W—�����صĺ�������λΪ��ÿ�ٿ�����ÿ�ٺ���(g/100g��g/100mL)��
A1—����ʹ��Һɫ��������ߣ�
A2—��ʹ��Һ��������ߣ�
C—��ʹ��ҺŨ�ȣ���λΪ����ÿ����( mg/mL)��
V—���������������λΪ����( mL)��
m—�������������������λΪ�˻�����( g��mL)��
100—��λת����
1000—��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч������
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
8 ����
��������������״����������Ϊ0.1g���������Ϊ5.0mLʱ���������ļ����Ϊ0.20g/100g����Һ��ȡ����Ϊ0.2mL���������Ϊ10mLʱ���������ļ����Ϊ0.22g/100mL��
��¼A
����Һ��������Һ��������ɫ��ͼ
A.1����������Һɫ��ͼ����ͼA.1��
ͼA.1����������Һɫ��ͼ
A.2���д�������������Һɫ��ͼ����ͼA.2��

ͼA.2���д�������������Һɫ��ͼ
��������ʳƷ��«���յIJⶨ
Determination of aloin in health food
1 ��Χ
�����涨�˱���ʳƷ��«������Һ��ɫ�ײⶨ������
������������«������ӹ�ƷΪԭ�ϵı���ʳƷ��«���պ����IJⶨ��
2 ԭ��
��Ʒ�ü״�+ˮ��55+45����Ϊ�ܼ�����ȡ�����е�«���գ���Һ��ɫ����C18 �����룬��293 nm�������������«���ձ���ʱ�䶨�ԣ������������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 ����
3.1.1 �״�(CH3OH)��ɫ�״���
3.1.2 ʯ���ѣ��г�30����60����
3.2 ��Ʒ
«���ձ�Ʒ(C21H22O9)��
3.3 ����Һ������
3.3.1 «���ձ�������Һ����ȡ10.00 mg«���ձ�Ʒ��25 mL ����ƿ�������������ܽⲢ�������̶���ҡ����
3.3.2 «����������Һ���ֱ���ȡ«���ձ�������Һ0.5 mL��1.0 mL��2.0 mL��4.0 mL��6.0 mL��10 mL����ƿ�У�����������������Ũ��Ϊ20 μg/mL��40 μg/mL��80 μg/mL��160 μg/mL��240 μg/mL�ı�����Һ��
4 �����豸
4.1 ��ЧҺ��ɫ���ǣ���������������������м������
4.2 ��������ϴ����
4.3 ������ƽ������1 mg��0.1 mg��
4.4 ���Ļ���ת��≥3000 r/min��
4.5 ����Ĥ��0.45 μm��
5 ��������
5.1 �����Ʊ�
5.1.1 ��������
��ȡ�ѷ����Ͼ��ȵĴ�������1 g(��ȷ�� 0.001 g)���þ�����ƿ�У�����50.0 mL�����࣬��������������10 min�����䣬�������������ʧ��������ҡ�ȣ���0.45 μm����Ĥ��������Һ���⡣��Ҫʱ�ɽ����ʵ�ϡ�͡�
5.1.2���ͻ�������
��ȡ��Ͼ��ȵĴ�������1 g(��ȷ�� 0.001 g)���þ�����ƿ�У�����25.0 mLʯ���ѣ�����ʹ��ֻ��ȣ����ˣ���ȥʯ����Һ����������ʯ����ϴ����ƿ����ֽ���Ӹɣ�����ֽ�Ͳ�������ԭ������ƿ�У�����50.0 mL�����࣬��������������10 min�����䣬�������������ʧ��������ҡ�ȣ���0.45 μm����Ĥ��������Һ���⡣��Ҫʱ�ɽ����ʵ�ϡ�͡�
5.1.3 ˮ��Һ������
��ȡ������������Ҫʱ���������ʵ�ϡ�ͣ����ģ�ȡ����Һ��0.45 μm����Ĥ���ˡ�
5.2ɫ�ײο�����
5.2.1 ɫ������C18��������250 mm���ھ�4.6 mm������5 μm����ͬ�����ܵ�ɫ������
5.2.2�����ࣺ�״�+ˮ=55+45��
5.2.3���٣�1 mL/min��
5.2.4���£�40����
5.2.5��Ⲩ����293 nm��
5.2.6��������10 μL��
5.3 �����ߵ�����
����ϵ�и�Ũ����Һ(3.3.2)��ע��Һ��ɫ�����У������Ӧ�ķ�������Ա�����Һ��Ũ��Ϊ�����꣬�Է����Ϊ�����꣬���Ʊ�����(����ҺҺ��ɫ��ͼ����¼A��ͼA.1 )��
5.4 ������Һ�IJⶨ
��������Һ(5.1.1��5.1.2��5.1.3)ע��Һ��ɫ���������Ա���ʱ�䶨�ԣ�ͬʱ��¼����������ݱ����ߵõ�����Һ��«���յ�Ũ��(��Ʒ��ҺҺ��ɫ��ͼ����¼A��ͼA.2)��
6 �������
������«����������ʽ(1)������
Ci×V ×100
Xi = ..........................................(1)
m×1000
ʽ�У�
Xi ������«��������������λΪ��ÿ�ٿ���g/100 g�����ÿ�ٺ�����g/100 mL����
Ci——�ɱ����߲�òⶨ��Һ��«������Ũ�ȣ���λΪ����ÿ������mg/mL����
V ���ⶨ��Һ�����ն����������λΪ������mL����
m ������������������λΪ�ˣ�g���������mL����
100——��λת����
1000——��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
8 ����
��������Ϊ1 g���������Ϊ50 mLʱ��«�����Ķ�����Ϊ0.0046g/100g��
��¼ A
����Һ��������Һ����Һ��ɫ��ͼ
A.1 «��������Һɫ��ͼ����ͼ A.1��
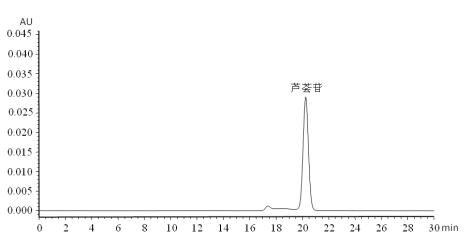
ͼ A.1 «��������Һɫ��ͼ
A.2 ����«������������Һɫ��ͼ����ͼ A.2��
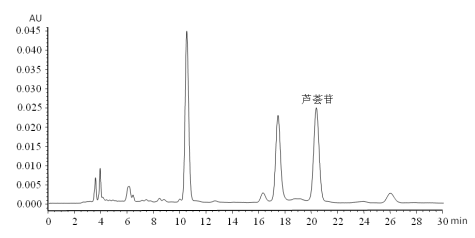
ͼ A.2 ����«������������Һɫ��ͼ
�ġ�����ʳƷ�����IJⶨ
Determination of carnitine in health food
1��Χ
�������涨�˱���ʳƷ������Һ��ɫ�ײⶨ������
�����������������Ϊ��Ҫԭ�ϵı���ʳƷ�����IJⶨ��
2ԭ��
�����е������0.5mmol/L��������Һ��������ȡ������ɫ���룬�Ա�Ʒ�ı���ʱ�䶨�ԣ���귨������
3�Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪGB/T 6682�涨��һ��ˮ��
3.1 �Լ�
3.1.1 ���������(K2HPO4)��
3.1.2 ���������(C8H19NaO3S)��
3.1.3 ����(HCl)��
3.1.4 ����(H3PO4)��
3.1.5������(SiO2)����ѧ����
3.1.6����(C2H3N)��ɫ�״���
3.2 �Լ�����
3.2.1 ������Һ(0.50 mmol/L)����ȡ4.2mL���ᣬ��ˮϡ�Ͳ�������100mL������ȡ������Һ1mL����ˮϡ�Ͳ�������1L��
3.2.2 ��������Һ���ֱ��ȡ8.7g��������غ�0.4325g��������ƣ���ˮ�ܽⲢϡ����1L�����������pH2.5��
3.3 ��Ʒ
���(C7H15NO3)������≥98%��������֤�����������֤��ı����ʡ�
3.4 ����Һ����
3.4.1 ��������Һ(1.0mg/mL)�����ܳ�ȡ����Ʒ25 mg����ȷ��0.1mg����25 mL����ƿ�У���������Һ�ܽⲢ�������̶ȣ�ҡ�ȡ�
3.4.2 ��������Һ���ֱ�ȷ��ȡ���������Һ0.50 mL��1.0 mL��2.0 mL��3.0 mL��4.0 mL��5.0 mL��5 mL����ƿ������������Һϡ�����̶ȣ���Ũ��Ϊ0.10 mg/mL��0.20 mg/mL��0.40 mg/mL��0.60 mg/mL��0.80 mg/mL��1.0 mg/mL�ı�����Һ��
4����
4.1 ��ЧҺ��ɫ���ǣ�����������UV�����������������У�DAD���������
4.2 ��ƽ������ 0.1mg��0.01mg
4.3 ��������ȡ����
5��������
5.1 �����Ʊ�
5.1.1 ��������
5.1.1.1 ��������
��ȡ���鲢��Ͼ��ȵ�����0.1 g~2 g (��ȷ��0.001g)(���������Լ5 mg~50 mg)����50mL����ƿ�У�����������ҺԼ35mL��������ȡ10min�����䣬��������Һϡ�����̶������ȣ����ˣ�������Һ���������ռ���Һ����0.45μmˮ����Ĥ��ȡ����Һ������
5.1.1.2 ����������
ȡ�������������������������ȣ���ȡ0.1 g~2 g(��ȷ��0.001g)�����������������������ɢ���ȣ���ȡ���в���(��ȷ��0.001g�����������Լ5 mg~50 mg)��ת����250 mL��������ƿ�У�����ȡ������Һ50mL����������ƿ�У����أ�����������ȡ10 min�����䣬��������Һ�������������ȣ����ˣ�������Һ���������ռ���Һ����0.45 μmˮ����Ĥ��ȡ����Һ���⡣
5.1.1.3 Һ������
������ȡ���Ⱥ������1.0 mL~5.0 mL (���������Լ5 mg~50 mg)����50 mL����ƿ��������������ҺԼ35 mL��������ȡ10 min�����䣬��������Һϡ�����̶������ȣ����ˣ�������Һ���������ռ���Һ����0.45 μmˮ����Ĥ��ȡ����Һ������
5.1.2 ϡ��
����������������������Һ�����ʵ���ϡ�ͣ�F����ʹ������Һ������Ũ����0.10 mg/mL~ 1.0 mg/mL��
5.2 ɫ�ײο�����
5.2.1 ɫ������C18����4.6×250mm��5μm��ͬ�����ܵ�ɫ������
5.2.2 �����ࣺ��������Һ+�Ҿ�=90+10��
5.2.3 ���٣�0.8mL/min��
5.2.4 ��Ⲩ����210nm��
5.2.5 ��������20μL��
5.3 �����ߵ�����
����������Һ(3.4.2)�ӵ�Ũ�ȵ���Ũ�ȷֱ�ע���ЧҺ��ɫ�����У��ⶨ��Ӧ�ķ�������Ա�����Һ��Ũ��Ϊ�����꣬�Է����Ϊ�����꣬���Ʊ�����(����ҺҺ��ɫ��ͼ����¼A��ͼ A.1 )��
5.4������Һ�IJⶨ
����ͬɫ�������£���������Һ(5.1.1.1��5.1.1.2��5.1.1.3��5.2.1)ע���ЧҺ��ɫ�����У��Ա���ʱ�䶨�ԣ���÷���������ݱ������������Һ������Ũ��(������ҺҺ��ɫ��ͼ����¼A��ͼA.2)��
6 ��������ı���
������������ʽ(1)���㣺

ʽ�У�
X——���������ĺ�������̬������λΪ��ÿ�ٿ�(g/100g)��Һ̬������λΪ��ÿ�ٺ���(g/100mL)��
ρ——���ݱ������������Һ������Ũ������λΪ����ÿ����(mg/mL)��
V——����ϡ���������λΪ����(mL)��
m——����ȡ����������λΪ��(g)������ȡ���������λΪ����(mL)��
F——ϡ�ͱ�����
100——��λת����
1000——��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ���ó�������ƽ��ֵ��5%��
8 ����
��������Ϊ2.0 g���������Ϊ50mLʱ�����Ķ�����Ϊ0.10 g/100g��
��ȡ����Ϊ5.0mL���������Ϊ50mLʱ�����Ķ�����Ϊ0.040 g/100mL��
��¼ A
����Һ��������Һ����Һ��ɫ��ͼ
A.1 �������Һɫ��ͼ����ͼ A.1��
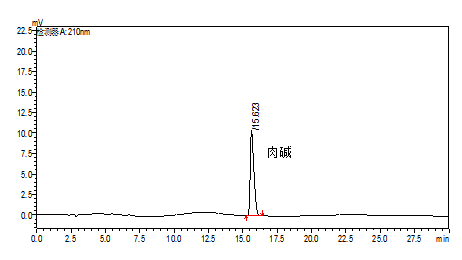
ͼA.1 �������Һɫ��ͼ
A.2 �������Ĵ�����Һɫ��ͼ����ͼA.2��
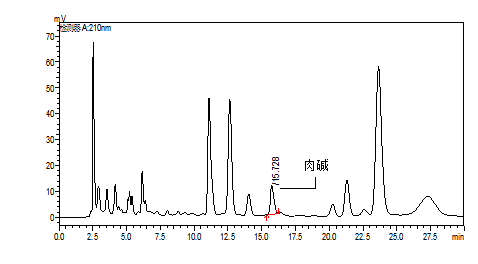
ͼA.2 ���������������Һɫ��ͼ
��¼ A
����Һ��������Һ����Һ��ɫ��ͼ
A.1 �������Һɫ��ͼ����ͼ A.1��

ͼA.1 �������Һɫ��ͼ
A.2 �������Ĵ�����Һɫ��ͼ����ͼA.2��
ͼA.2 ���������������Һɫ��ͼ
�塢����ʳƷ��α-�����ᡢγ-������IJⶨ
Determination of α, γ-linolenic acids in health food
1 ��Χ
�������涨�˱���ʳƷ��α-��γ-������IJⶨ������
��������������֬������ʳƷ��α-��γ-�����Ậ���IJⶨ��
����������������֬������ʳƷ��C16~C22������֬����ͽ���ϩ�����IJⶨ��
2 ԭ��
����֬����(��������ȡ��֬��)��������������������������������¼״�������Ȼ��������ɫ���Ƿ�����������귨������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1��������C6H14����
3.1.2 ��������( KOH)��
3.1.3��������������Һ��
3.1.4 �״�(CH3OH)��ɫ�״���
3.1.5 �Ȼ���(NaCl)��
3.2 �Լ�����
3.2.1�������ؼ״���Һ (0.5 mol/L):��ȡ2.8 g�������أ��ü״��ܽⲢ������ 100 mL�����ȡ�
3.2.2��������״���Һ(1+4)��ȡ40%��������������Һ1�ݣ��Ӽ״�4�ݣ����ȼ��ɡ�
3.2.3�����Ȼ��ƣ������Ȼ�����Һ:��ȡ360g�Ȼ����ܽ���1.0Lˮ��,�����ܽ�,���屸�á�
3.3.1 α-���������>99.0%��
3.3.2 γ-���������>99.0%��
3.3 ����Һ������
3.4.1 ������Һ(1.0 mg/mL)���ֱ���ȡ25.0mg α-�����������25.0mg γ-�����������Ʒ����25mL����ƿ�������������ܽⲢ�������̶���ҡ��������ҺӦ������-18°C �����С�
3.4.2 ��ʹ��Һ(0.5 mg/mL)���ֱ���ȡα-�����������γ-���������������Һ��5.0mL��10mL������ƿ�У����ȣ� ����ʱ���ơ�
4 �����豸
4.1����ɫ���ǣ��������(FID)�������
4.2������ƽ������0.1mg��1mg��
4.3����ʽ������������
4.4��ĥ����ƿ(50mL)��ֱ�������ܡ�
5 ��������
5.1�����Ʊ�
5.1.1֬������ȡ ��GB 5009.6�й涨�ķ�����ȡ��
5.1.2����
��ȡ0.100g��֬(��֬��)�ʹ���������һ������50mLĥ����ƿ��(��ͼ1)������4mL0.5mol/L�������ؼ״���Һ���ϲ����ӻ��������ܣ����̶��ڴ����������ϣ����������Ͽ�����Һ�е��뵪����ʹ��Ӧƿ��ʼ�ճ�������������������������������ʹ��ӦҺ����65±5�����������Լ15min��
5.1.3��֬��
���������ϲ�����4mL��������״���Һ������(65±5��)������Լ2min���������£� ���������ϲ�����5mL�������������5min����ȥ�����ܣ�����5mL�����Ȼ���ˮ��Һ�� ҡ�������ӣ�ת����25mL��Һ©���з���ˮ���л��࣬�ټ�3mL������ϴˮ�࣬���룬��ˮ�࣬�ϲ��л��ಢ��������������10.0mL(Ũ�ȵ�ʱ����Ũ����1.0mL)�����ⶨ�á�
5.2����ɫ�ײο�����
5.2.1ɫ������FFAP(���Ծ��Ҷ���20M��30m×0.25mmi.d.0.25μm)
5.2.2 �����¶ȣ�215����
5.2.3�������¶ȣ�250����
5.2.4������¶ȣ�260����
5.2.5������50mL/min��30��1������������45mL/min��������500mL/min��
5.3���Է���
���������������£��ֱ�ȡ��ʹ��Һ�������ⶨҺ1.0μL��ע������ɫ���ǣ��Ա���ʱ����ȷ��α-��γ-�����������
5.4��������
������α-�����������γ-���������ɫ�������������ıȽ϶�����
6 ��������ı���
������α-�����������γ-������ⶨ�����(1)ʽ����
6.1����

..........................................(1)
ʽ����
X—α-�������γ-�����Ậ����%��
A1—������α-�����������γ-���������ɫ��������ߣ�
A2—��ʹ��Һɫ��������ߣ�
ρ—��ʹ��ҺŨ�ȣ�mg/mL��
v—�����鶨�������mL��
m—����������g��
0.952—�����ỻ��ϵ����
֬�������ٻ���ԭ����ʳƷ������γ-�������α-�����������
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
8 ����
��������ͼ������γ-������Ϊ0.050μg��α-������Ϊ0.030μg��
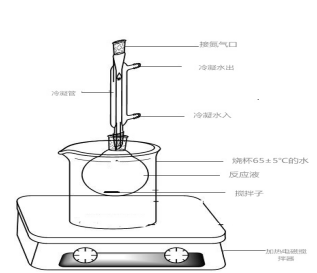
ͼ 1��������װ��ͼ
��¼ A
����Һ��������Һ��������ɫ��ͼ
A.1 α-�������������Һɫ��ͼ����ͼ A.1��

ͼ A.1 γ-��������� α-�������������Һɫ��ͼ
A.2 ����α-�����������������Һɫ��ͼ����ͼ A.2��

ͼ A.2 ����α-�����������������Һɫ��ͼ
��������ʳƷ���˲����յIJⶨ
Determination of ginsenosides in health food
1 ��Χ
�������涨�˱���ʳƷ���˲����յĸ�ЧҺ��ɫ�ײⶨ������
���������������˲μ���ӹ�ƷΪ��Ҫԭ�ϵı���ʳƷ���˲�����Re��Rg1��Rb1��Rc��Rb2��Rd�����IJⶨ��
2 ԭ��
�������е��˲������ܽ⡢��ȡ��������������ʹ���ݶ�ϴ�ѷ����ЧҺ��ɫ���з��룬����������⣬����ɫ��ı���ʱ�䶨�ԣ���귨������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪGB/T 6682�涨��һ��ˮ��
3.1 �Լ�
3.1.1 ����(CH3CN)��ɫ�״���
3.1.2 �״�(CH3OH)��
3.1.3 D101���������֬��
3.2 ��Ʒ
�˲�����Re��Rg1��Rb1��Rc��Rb2��Rd������≥98%��������֤�����������֤��ı����ʡ�
3.3 ����Һ������
3.3.1 �˲�����Re��Rg1��Rb1��Rc��Rb2��Rd������Һ(10mg/mL)���ֱ��ȡ�˲�����Re��Rg1��Rb1��Rc��Rb2��Rd��Ʒ100mg��6��10.0mL����ƿ�У��ü״��ܽⲢ�������̶ȣ�ҡ�ȡ�
3.3.2 �˲�����Re��Rg1��Rb1��Rc��Rb2��Rd��ϱ�����Һ���ֱ���ȡ�˲�����Re��Rg1��Rb1��Rc��Rb2��Rd������Һ0.10mL��0.20mL��0.40mL��0.80mL��1.00mL��10.0mL����ƿ�У��ü״����ݣ���Ũ��Ϊ0.10mg/mL��0.20 mg/mL��0.40 mg/mL��0.80 mg/mL��1.00 mg/mL�Ļ�ϱ�����Һ��
4 �����豸
4.1 ��ЧҺ��ɫ���ǣ�������������
4.2 ��������ϴ����
4.3 ���Ļ���
4.4 ˮԡ����
4.5 ������ƽ������1mg��0.1mg��
5 ��������
5.1 �����Ʊ�
5.1.1 ��������
ȡ�����гɷ�ĩ������20Ŀɸ����ȡ�÷�ĩ���������൱�ں����˲�����Լ75mg����ȷ��0.001g������50mL����ƿ�У���ˮ45mL�ڳ�������ϴ���г�����ȡ30���ӣ�ȡ�������������º�ˮ�������̶ȣ�ҡ�ȣ��˹���ȷ��ȡ����Һ10mL��ͨ��D101���������֬�����������������֬ʹ��ǰ�Ⱦ��״����ݣ�ˮϴ��װ��10cm����ֱ��1~1.5 cm��С������С������10mLˮ��ϴ����ȥˮҺ֮����70%�״�25mLϴ�����գ��ռ��״���Һ��ˮԡ�����ɣ������Լ״��ܽⲢ������5.0mL������Һ���ĺ��0.45μmĤ����Һ����ɫ������
5.1.2 Һ������
ȡһ�������������൱�ں����˲�����Լ75mg������ˮԡ�����ɣ�������50mLˮ������ȡ30���ӣ����²�����5.1.1��ͬ��
5.2 ɫ�ײο�����
5.2.1 ɫ������ C18����4.6 x 250 mm��5μm��ͬ�����ܵ�ɫ������
5.2.2��Ⲩ�� 203nm��
5.2.3 ���£�35°C��
5.2.4 ��������5μl��
5.2.5 �����ࣺA��Ϊ���棬B��Ϊˮ��
�ݶ�ϴ������
|
ʱ�䣨min��
|
A�ࣨ����
|
B�ࣨ����
|
���٣�mL/min��
|
|
0
|
16
|
84
|
1.0
|
|
20
|
18
|
82
|
1.0
|
|
55
|
40
|
60
|
1.0
|
|
65
|
40
|
60
|
1.0
|
|
66
|
100
|
0
|
1.0
|
|
71
|
100
|
0
|
1.0
|
|
72
|
16
|
84
|
1.0
|
|
85
|
16
|
84
|
1.0
|
5.3 �����ߵ�����
��5μL�Ļ�ϱ�����Һע��Һ��ɫ�����У��ⶨ��Ӧ�ķ�������ߣ��Ա�����Һ��Ũ��Ϊ�����꣬�Է��������Ϊ�����꣬���Ʊ�����(����ҺҺ��ɫ��ͼ����¼A)��
5.4 ������Һ�IJⶨ
��5μL����������Һע��Һ��ɫ�����У��Ա���ʱ�䶨�ԣ���÷�������ߣ����ݱ����ߵõ�����Һ�˲�����Re��Rg1��Rb1��Rc��Rb2��Rd��Ũ�ȡ�
6 ��������ı���
�����и��˲����յĺ�����ʽ(1)���㣺
ʽ�У�
Xi——���������˲����յĺ�������λΪ��ÿ�ٿ����ÿ�ٺ���(g/100g��g/100mL);
Ci——�ɱ����߲�òⶨ��Һ�����˲�������Ũ�ȣ���λΪ����ÿ����(mg/mL);
V——���ⶨ��Һ�����ն����������λΪ����(mL);
F——���ⶨ��Һ��ϡ��������
m——������ȡ��������λΪ�˻�����(g��mL);
100——��λת��;
1000——��λת����
���������˲����յĺ�����ʽ(2)���㣺
X����XRe��XRg1��XRb1��XRc��XRb2��XRd………………(2)
ʽ�У�
X��—���������˲����յĺ�������λΪ��ÿ�ٿ����ÿ�ٺ���(g/100g��g/100mL);
Xi—�����и��˲����գ�Xi�����˲�����Re��Rg1��Rb1��Rc��Rb2��Rd���ĺ�������λΪ��ÿ�ٿ����ÿ�ٺ���(g/100g��g/100mL)��
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ��10%��
8 ����
�������������˲����յ���ͼ����Ϊ10 mg/kg��
�������������˲����յ�������Է�Χ��0.1mg/mL��1mg/mL��
��¼A
����Һ����Һ��ɫ��ͼ
A.1 �˲�����Rg1��Re��Rb1��Rc��Rb2��Rd�ı���Һɫ��ͼ����ͼA.1��

ͼA.1 �˲�����Rg1��Re��Rb1��Rc��Rb2��Rd�ı���Һɫ��ͼ
A.2 �˲�����Rg1��Re��Rb1��Rc��Rb2��Rd����Ʒ��Һɫ��ͼ����ͼA.2��

ͼA.2 �˲�����Rg1��Re��Rb1��Rc��Rb2��Rd����Ʒ��Һɫ��ͼ
�ߡ�����ʳƷ��ԭ�����صIJⶨ
Determination of procyanidins in health food
1 ��Χ
�������涨�˱���ʳƷ��ԭ�����صIJⶨ������
�����������ڱ���ʳƷ��ԭ�����صĺ����ⶨ��
2 ԭ��
ԭ�������Ǻ��ж����غͱ������ص�Ԫ�ľۺ��ԭ�����ر�����ɫ�����������ᴦ�������������ɫ�Ļ��������ӡ������÷ֹ��ȷ��ⶨԭ��������ˮ����������ɵĻ��������ӡ�����������ԭ�����غ�����
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1 �״���CH3OH����
3.1.2 ��������CH3(CH2)3OH����
3.1.3 ������HCl����
3.1.4 �������(NH4Fe��SO4��2·12H2O)
3.2 �Լ�����
3.2.1������2 mol/L����ȡ����90mL����ˮ����ʹ��500mL��ҡ�ȡ�
3.2.2 ���������Һ����ȡ10g������泥���2 mol/L�����ܽⲢ������500mL�����ȣ�����Һ���������Ũ��Ϊ2%��w/v����
3.3 ��Ʒ
ԭ����������������Դ��������≥95%��
3.4 ��Ʒ��Һ������
ԭ������������Һ(1.0 mg/mL)����ȡ10 mg(��ȷ��0.1 mg)ԭ�����ر�Ʒ��10 mL ����ƿ�У��ü״��ܽⲢ�������̶ȣ�ҡ�ȡ�
4 ����
4.1 ��ƽ������Ϊ 1 mg �� 0.1 mg��
4.2 �ֹ��ȼơ�
4.3 ���Ļ���ת�� ≥4000 r/min��
4.4 �����ǡ�
4.5 ����װ�á�
5 ��������
5.1 �����Ʊ�
5.1.1 ������������ȡ�ѷ����Ͼ��ȵĴ�������50 mg~100 mg(��ȷ��0.1 mg)������50 mL����ƿ�У�����30 mL�״�����������20 min�����������ºӼ״����̶ȣ�ҡ�ȣ����Ļ�����������ȡ����Һ���á�
5.1.2 ������������ȡ��Ͼ��ȵĴ�������50 mg(��ȷ��0.1 mg)������С�ձ��У���20 ��30mL�״������ν��裬����ȡҺת����50 mL����ƿ�У�ֱ���״���ȡҺ��ɫ���Ӽ״����̶ȣ�ҡ�ȡ�
5.1.3 Һ����������ȡ������1 mL�Ĵ�������������50 mL����ƿ�У��Ӽ״����̶ȣ�ҡ�ȡ�
5.2 �����ⶨ
�������������ᰴ95:5������Ȼ�Ϻ�ȡ��6.0 mL���ھ�����ƿ�У��ټ���0.2 mL���������Һ��1.0 mL������Һ�����ȣ��÷�ˮԡ��������ȷ����40 min�������ñ�ˮ����ȴ���ڼ������15 min����546 nm������������ȣ��ɱ�������������ԭ�����صĺ�������ɫ��1Сʱ���ȶ���
5.3 �������Ʊ�
�ֱ���ȡԭ������������Һ0 mL��0.1 mL��0.25 mL��0.5 mL��1.0 mL��1.5 mL����10 mL����ƿ�У��Ӽ״����̶ȣ�ҡ�ȡ�����ȡ1.0mL�ⶨ���������ⶨ������ͬ������ԭ������Ũ��������ȹ�ϵ�ı����ߡ�
6 �����������
������ԭ�����زⶨ�������1��ʽ����
C×V×1000
X= ———————————×100………………..��1��
m×1000×1000
ʽ�У� X—������ԭ�����صĺ����� g/100g��
C—�ɱ������ϲ������������ԭ��������Ũ�ȣ�μg/mL��
V—������������������� mL��
m—����������g��
�������������λ��Ч���֡�
7 ��������
���ܶȣ����ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ��10%��
8 ����
��������������0.2mg/g��
�ˡ�����ʳƷ�к�����IJⶨ
Determination of nucleotide in health food
1 ��Χ
�������涨�˱���ʳƷ�к�����ĸ�ЧҺ��ɫ�ײⶨ������
����������ޣ�����ऺ��գ�CMP��0.04μg/ mL������ऺ��գ�UMP��0.05μg/ mL�������ʺ��գ�AMP��0.05μg/ mL�������ʺ��գ�GMP��0.06μg/ mL��
���������Է�Χ������ऺ��գ�CMP��0.992��12.4μg/ mL������ऺ��գ�UMP��1.17��14.6μg/ mL�������ʺ��գ�AMP��1.01��12.6μg/ mL�������ʺ��գ�GMP��0.948��12.3μg/ mL��
2 ԭ��
�������ܽ⡢ȥ������ʹ�ð���������ȡ���Ժ�������о������������ݸ�ЧҺ��ɫ������������254nm������Ӧ���ж��Զ�����
3 �Լ�
ע��������˵���������Լ���Ϊ��������ʵ����ˮ����GB/T 6682-2008һ��ˮҪ��
3.1 �״���CH3OH�����ż�����
3.2 �����C2H4O2ӦΪ36%��37%��g/g����
3.3 ����������KH2PO4����
3.4 �����������K2HPO4����
3.5 ����ι�����ȡ����
3.6 �������������Һ��ȷ������100������4h�����ı�Ʒ����ऺ��գ�CMP��100mg������ऺ��գ�UMP���������ʺ��գ�AMP���������ʺ��գ�GMP����40mg��100mL����ƿ�У���ˮ�ܽⲢ�������̶� ��
3.7 �������ʹ��Һ�����������������Һ��0.25mol/L��pH=3������������Һϡ��100����
4 �����豸
4.1 ��ƽ������Ϊ 1 mg �� 0.1 mg��
4.2 ��ЧҺ��ɫ���ǣ������������л�������������
5 ��������
5.1 ��������
5.1.1 ��ȡ�����������൱�ں�������Լ6.5mg����ȷ��0.001g������100mL����ƿ�У�����Լ50����ˮ80mL�������ȡ�������Һ�ﵽ���º���ˮ�������̶ȡ�
5.1.2 ȷ��ȡ10mL������Һ��100mL����ƿ�У�����0.5������5mL��ˮ10mL�����Ⱥ���5min�Գ������ס���ˮ�������̶ȣ����Ⱥ���ˣ���ȥ����������Һ���ռ�Լ30mL��Һ��
5.1.3 ������ι�����ȡ������10mL�״���10mLˮ����ٽ�20mL������Һ���ˣ���1mLˮ��ϴ��ȡ������0.25mol/L��pH=3.5����������Һϴ�ѳ�5mL��Һ��ȫ��������Ҫ��������1��/�롣
5.2 Һ��ɫ�ײο�����
5.2.1 ɫ����: C18 �� 3.9×150mm��5μm��ͬ������ɫ����
5.2.2 ���£�25����
5.2.3��Ⲩ�� 254nm��
5.2.4 �����ࣺ0.01mol/L��������—0.1mol/L��������أ�480��20��
5.2.5 ���٣�0.6mL/min��
5.2.6 ��������10mL��
5.2.7ɫ��������ȡ10μL����Һ��������Һע��ɫ�����У��Ա���ʱ�䶨�ԣ������������������Ƚ϶�����
5.3 �����ߵ��Ʊ����ֱ�����Ũ��Ϊ1.0��2.0��5.0��10.0��16.0μg/mL��4�ֺ��������Һ���ڸ��������������½���Һ��ɫ�������Է��������Ũ���������ߡ�
6���������
ʽ�У�
X—�����к�����ĺ�����mg/100g��
h1—�������������
C—����ҺŨ�ȣ�μg/mL��
K—ϡ�ͱ�����
h2—����Һ���������
m—��������g��
�������ܺ�����ĺ���Ϊ����ऺ��գ�CMP��������ऺ��գ�UMP���������ʺ��գ�AMP���������ʺ��գ�GMP������֮�͡�
�����������λ��Ч���֡�
7����������
���ظ��������£���õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ��10%��
��������ʳƷ���工��͡�������ⶨ
Determination of lovastatin in health food
1 ��Χ
�������涨�˱���ʳƷ���工��͡��Һ��ɫ�ײⶨ������
�������������Ժ�������ӹ�ƷΪԭ�ϵı���ƷʳƷ���������ջ����工��͡����ʽ���������工��͡�IJⶨ��
2 ԭ��
������75% �Ҵ���Һ������ȡ������Һ��ɫ�����������ջ�������ʽ���������工��͡������������⣬�Ա���ʱ�䶨�ԣ���귨������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1 �״�(CH3OH)��ɫ�״���
3.1.2 ��ˮ�Ҵ�(CH3CH2OH)��
3.1.3 ����(H3PO4)��
3.1.4 �������ƣ�NaOH��
3.1.5 ���ᣨHCl������������Լ37 %��
3.2 �Լ�����
3.2.1 75% �Ҵ�(v/v)������ˮ�Ҵ���ˮ��75+25������Ȼ�Ͼ�����
3.2.2 0.2 mol/L����������Һ����ȡ��������1.6g����ˮʹ�ܽ��200ml�����á�
3.2.3 0.2 mol/L������Һ����ȡ����1.8ml����ˮ����ʹ��100ml�����á�
3.3 ��Ʒ
�工��͡(C24H36O5)��CAS���75330-75-5������≥98%��
3.4 ����Һ������
3.4.1 �工��͡������Һ(400 μg/mL)��ȷ��ȡ�工��͡��Ʒ40 mg����ȷ��0.01 mg������75% �Ҵ��ܽⲢ������100 mL��
3.4.2 �工��͡��ϵ��������Һ����������ϡ��������Ũ��Ϊ 8.0 μg/mL��20 μg/mL��40 μg/mL��80 μg/mL��160 μg/mL��320 μg/mL ��ϵ�б�����Һ��
3.4.3 ��������ʽ��������������͡��Һ����ȡ�工��͡����������Ʒ4mg����0.2mol/L����������Һ������100 mL����50�������³���ת��1h�����õ����º��ٷ���1h����0.2mol/L ������Һ����pH�����ԡ�
4 �����豸
4.1 ��ЧҺ��ɫ�����������������л�����������UV����
4.2 ������ƽ������Ϊ 1 mg �� 0.01 mg��
4.3 ��������ϴ����
4.4 �����������
4.5 ���Ļ���ת��≥3500 r/min��
5 ��������
5.1 �����Ʊ�
���������鲢��Ͼ��ȣ���ȡ�����������൱�ں��工��͡Լ5 mg����ȷ��0.001g�����þ�����ƿ�У���������50 mL 75%�Ҵ����������ƶ�������������ȡ60 min���������ٳƶ���������75% �Ҵ������ʧ��������ҡ������3500 r/min ��ת������10 min��ȡ����Һ����0.45 μm����Ĥ���ˣ���Һ��Ϊ��������Һ��
5.2ɫ�ײο�����
5.2.1 ɫ������C18����4.6×250 mm��5 μm��ͬ������ɫ������
5.2.2 ���£�������
5.2.3 ��Ⲩ����238 nm��
5.2.4 ���������״�+ˮ+����=385+115+0.14��
5.2.5 ���٣�1.0 mL/min��
5.2.6 ��������10 μL��
5.3 �����ߵ�����
��10 μL����ϵ�й���Һ�ֱ�ע��Һ��ɫ�����У������Ӧ�ķ�������Ա�ϵ�й���Һ��Ũ��(μg/mL)Ϊ�����꣬�Է����Ϊ�����꣬���Ʊ����ߡ�
5.4 ������Һ�IJⶨ
��10 μL ��������Һע��Һ��ɫ�����У��Ա���ʱ�䶨�ԣ��ⶨ������ַ���������ݱ����ߵõ�����Һ�������ͣ��ջ����工��͡����ʽ���������工��͡��Ũ��(μg/mL)��
6 ��������ı���
6.1 ���������������ջ����工��͡����ʽ���������工��͡�������ֱ�ʽ(1)������

ʽ����
Xi——�����������ͣ��ջ����工��͡����ʽ���������工��͡�ĺ�������λΪ��ÿ�ٿ�(g/100g)��
Ci——���ⶨ��Һ�������ͣ��ջ����工��͡����ʽ���������工��͡��Ũ�ȣ���λΪ��ÿ����(μg/mL)��
V——���ⶨ��Һ�����ն����������λΪ����( mL)��
m——�����ij�����������λΪ��( g)��
100——��λת����
1000——��λת����
6.2 ���������������ջ����工��͡����ʽ���������工��͡����������ʽ(2)������

ʽ����
X——�����������ͣ��ջ����工��͡����ʽ���������工��͡���ܺ�������λΪ��ÿ�ٿ�(g/100g)��
X1——������������(�ջ�)�工��͡����������λΪ��ÿ�ٿ�(g/100g)��
X2——��������ʽ(����)�工��͡����������λΪ��ÿ�ٿ�(g/100g)��
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
8 ����
��������Ϊ 0.5 g���������Ϊ 50 mLʱ���工��͡�Ķ�����Ϊ0.040 g/100g��
��¼A
����Һ��������Һ����Һ��ɫ��ͼ
A.1�����ͣ��ջ����工��͡����Һɫ��ͼ����ͼA.1��

ͼA.1 �����ͣ��ջ����工��͡����Һɫ��ͼ
A.2��ʽ���������工��͡����Һɫ��ͼ����ͼA.2��

ͼA.2 ��ʽ���������工��͡����Һɫ��ͼ
A.3�����工��͡��������Һɫ��ͼ����ͼA.3��

ͼA.3 �����工��͡��������Һɫ��ͼ
ʮ������ʳƷ��ֲ���Ч�ɷּ������鷽��
Distinguish method of plant function component in health food
��Χ
�������涨�˱���ʳƷ���˲Ρ�����Ρ���Ƥ���ʲݡ���ơ�����Ҷ��«���������ֲ���Ч�ɷֵļ�����
�����������ڱ���ʳƷ���˲Ρ�����Ρ���Ƥ���ʲݡ���ơ�����Ҷ��«���������ֲ���Ч�ɷֵļ���
һ ��Ƥ
1 ����Һ�Ʊ�
1.1 Ƭ������ ��ȡ��ϸ������2 g���Ӽ״�20 mL��ˮԡ�ϻ���20 min��������15 min���˹���ȡ��Һ10 mL��Ũ�����ɣ������Ӽ״�1 mLʹ�ܽ�����Ϊ����Ʒ��Һ��
1.2�ǿ飺ȡ��Ʒ30 g����30 mLˮʹ�ܽ⣬��ϡ�������pH��5��6����ˮ���͵���������ȡ3�Σ�25 mL��25 mL��20 mL�����ϲ���������ȡҺ����ˮϴ�����Σ�30 mL��30 mL����ȡ������Һ��1/3����ˮԡ�����ɣ������Ӽ״�1 mLʹ�ܽ⣬��Ϊ����Ʒ��Һ��
2 ����Һ�Ʊ�
ȡ��Ƥ����ҩ��0.3 g��ͬ����Һ�Ʊ�1.1���µķ����������Ƴɶ���ҩ����Һ����ȡ��Ƥ�ն���Ʒ�������Ӽ״��Ƴɱ�����Һ����Ϊ����Ʒ��Һ��
3 ���������0.5%����������Һ�Ʊ����轺G�������
4 ����������Һ����2μL��4 μL������Ʒ����2 μL��
5 չ��������1����������-�״�-ˮ��100:17:13����2���ױ�-��������-����-ˮ��20:10:1:1�����ϲ���Һ
6 չ����ʽ������չ��������չ������1������չ��3 cm��ȡ��������������չ������2������չ��8 cm��ȡ�������ɡ�
7 ��ɫ; ����3%���Ȼ����Ҵ���Һ���Լ��ȴ��ɣ�����������365 nm���¼��ӡ�
 |
8 ɫ��ʶ��������Ʒɫ�׳��۲쵽��Ƥ��ӫ��ߵ��⣬����Ʒ�����ҩ������ͬ��λ��������ͬ��ɫ��ӫ���㡣��ɫ���в������Ե�3����-����ɫӫ�����ߵ㣬Ϊ��Ҫ����������
1 ���ÿ��������ν���
2 ��Ƥ����ҩ��
3 ��Ƥ��
�� ���
1 ����Һ�Ʊ�
ȡ��ϸ��Ƭ�����ҷ�ĩ0.1 g���Ӽ״�20mL����1 h���˹���ȡ��Һ5 mL�����ɣ�������ˮ10 mLʹ�ܽ⣬�ټ�����1mL���÷�ˮԡ�ϼ��Ȼ���30 min��������ȴ����������ҡ��ȡ2�Σ�ÿ��20 mL���ϲ�����Һ�����ɣ����������ȼ���1 mLʹ�ܽ�����Ϊ����Ʒ��Һ��
2 ����Һ�Ʊ�
ȡ��ƶ���ҩ��0.1 g��ͬ����Һ�Ʊ����µķ����������Ƴɶ���ҩ����Һ����ȡ��������Ʒ���Ӽ״��Ƴ�ÿ1 mL��1 mg����Һ����Ϊ����Ʒ��Һ��
3 ���������0.3%�ȼ���ά����Ϊ��ϼ��Ĺ轺H�������轺G�������
4 ������������4 μL��
5 չ������ʯ���ѣ�30��60�棩-��������-���ᣨ15:5:1�����ϲ���Һ
6 չ����ʽ������չ��8 cm��
7 ��ɫ; �����ƣ�365 nm���¼��ӣ����ð�������Ѭ�����Ӻ��չ��¼��ӡ�
 |
8 ɫ��ʶ���������ƣ�365 nm���¼��ӣ�����Ʒɫ�������������ҩ��ɫ����Ӧ��λ����������ͬ�������Ȼ�ɫӫ�����ߵ����������Ʒɫ����Ӧ��λ����������ͬ�ijȻ�ɫӫ��ߵ㡣�ð�������Ѭ���չ��¼������ߵ��Ϊ��ɫ��
1 ���м��ʽ�֬��
2 ��ƶ���ҩ��
�� �ʲ�
1 ����Һ�Ʊ�
1.1 ��������
1.1.1 Ƭ���������͵Ľ��� ��ȡ��ϸ������2 g�����ھ���ƿ�У�������40 mL����ˮԡ�ϼ��Ȼ���1 h���˹�����ȥ����Һ�������Ӽ״�30 mL�����Ȼ���1 h���˹�����Һ���ɣ�������ˮ40 mLʹ�ܽ⣬��ˮ���͵���������ȡ3�Σ�ÿ��20 mL���ϲ�������Һ����ˮϴ��3�Σ���ȥˮҺ��������Һ��ˮԡ�����ɣ������Ӽ״�5 mLʹ�ܽ�����Ϊ����Ʒ��Һ��
1.1.2 ���ͽ��� ��ȡ����2 g�������۵���ֽ�ϣ���ʯ���ѷ�����ϴȥ��֬���ٽ���ֽ�ϵ�����ת�������ƿ�У���1.1.1������“������40 mL”��������������
1.1.3 �ǿ� ȡ������������ˮ40 mLʹ�ܽ�����1.1.1������“��ˮ���͵���������ȡ3��”��������������
1.1 Һ������
ȡ��10 mL����ˮϡ����40 mL����1.1.1������“��ˮ���͵���������ȡ3��”��������������
2 ����Һ�Ʊ�
ȡ�ʲݶ���ҩ��1 g��ͬ����Һ�Ʊ�1.1.1���µķ����������Ƴɶ���ҩ����Һ����ȡ�ʲ��ᵥ��ζ���Ʒ���Ӽ״��Ƴ�ÿ1 mL��2 mg����Һ����Ϊ����Ʒ��Һ��
3 ���������1%����������Һ�Ʊ��Ĺ轺G����塣
4 ��������1 μL��2 μL��
5 չ��������������-�״�-������-ˮ��15:1:1:2��
6 չ����ʽ��չ������չ����Ԥƽ��15 min������չ����չ��8 cm��
7 ��ɫ; ���������Ҵ���1→10����105 ���������ߵ���ɫ�������������ƣ�365 nm���¼��ӡ�
8 ɫ��ʶ��������Ʒɫ�������������ҩ��ɫ����Ӧ��λ����������ͬ��ɫ��ӫ��ߵ����������Ʒɫ����Ӧ��λ����������ͬ�ijȻ�ɫӫ��ߵ㡣
 |
1 ӥ�ƴ���������
2 �ʲݶ���ҩ��
3 �ʲ���
�� �˲Ρ������
1 ����Һ�Ʊ�
1.1 ��������
1.1.1Ƭ���������͵Ľ��� ��ȡ��ϸ������2 g�����ھ���ƿ���������ȼ���40 mL����ˮԡ�ϼ��Ȼ���1 h����ȥ���ȼ���Һ�������Ӹ��ܼ�����ˮ0.5 mL����ʪ��ˮ���͵�������10 mL����������30 min����ȡ����Һ���Ӱ���Һ��400→1000��3������ҡ�ȣ����÷ֲ㣬ȡ�ϲ�Һ���ɣ������Ӽ״�1 mLʹ�ܽ⣬��Ϊ����Ʒ��Һ��
1.1.2���ͽ��� ��ȡ����2 g�������۵���ֽ�ϣ���ʯ���ѷ�����ϴȥ��֬���ٽ���ֽ�ϵ�����ת�����ƿ�У���“�����ȼ���40 mL”��������������
1.2 Һ������
ȡ����10 mL����ˮ���͵�������20 mL����ҡ��ȡ����ȡ�������㣬�Ӱ���Һ��400→1000��3������ҡ�ȣ����÷ֲ㣬ȡ�ϲ�Һ���ɣ������Ӽ״�1 mLʹ�ܽ⣬��Ϊ����Ʒ��Һ��
2 ����Һ�Ʊ�
ȡ�˲λ�����ζ���ҩ��Լ1 g��ͬ����Һ�Ʊ�1.1.1���µķ����������Ƴɶ���ҩ����Һ����ȡ�˲�����Rb1��Re��Rg1��F11����Ʒ���Ӽ״��Ƴ�ÿ1 mL����2 mg�Ļ����Һ����Ϊ����Ʒ��Һ��
3 ��������轺G����塣
4 ����������Һ2 μL��10 μL������Һ2 μL��
5 չ���������ȼ���-��������-�״�-ˮ��15:40:22:10��10 �����·��õ��²�Һ��
6 չ����ʽ��չ�����ڼ���չ����10 mL������������������ʪ��Ϊ42%������57 mL+ˮ100 mL��������25 ����ƽ��15 min������չ��12 cm��14 cm��ȡ����������
7 ��ɫ; ���������Ҵ���Һ��1→10������105 ���������ߵ���ɫ�������ֱ����չ�������ƣ�365 nm���¼��ӡ�
8 ɫ��ʶ��������Ʒ�Ŀɼ���ɫ��ӫ��ɫ�������ҩ��ɫ�����������������κ��е��˲�����F11���˲β��������˲κ��е��˲�����Rf ������β���������Ϊ�˲�������μ��������������Ʒ�ߵ����¶�������ΪRb1��Re��Rg1��F11���˲�����Rfλ���˲�����Rg1��F11֮����

1. ���κ�Ƭ�����˲Ρ�����Σ� 1. �������������ң����˲Σ�
2. �˲ζ���ҩ�� 2. �˲ζ���ҩ��
3. ����ζ���ҩ�� 3. �˲�����Rb1��Re��Rg1��F11
�� ����Ҷ
1 ����Һ�Ʊ�
1.1 Ƭ���������͵Ľ��� ��ȡ��ϸ�ɷ�ĩ������4 g������250 mL����ƿ�У���50 %�ı�ͪ100 mL�����Ȼ���2.5 h���������˹�����Һ��ȥ��ͪ�����䣬��Һ������������ҡ��ȡ2�Σ�ÿ��50 mL���ϲ���ȡҺ��������������15%�Ҵ�2 mLʹ�ܽ⣬���ھ��������ϣ�30��60Ŀ��2.5 g�����ھ�1.5 cm���ɷ�װ��������5%�Ҵ�200 mLϴ�ѣ��ռ�ϴ��Һ��Ũ����50 mL��������������������ȡ2�Σ�ÿ��50 mL���ϲ���ȡҺ�����ɣ������ӱ�ͪ5 mLʹ�ܽ⣬��Ϊ����Ʒ��Һ��
1.2 ���ͽ��� ��ȡ����������4 g�������۵���ֽ�ϣ���ʯ���ѷ�����ϴȥ��֬���Ӹ��ܼ�������ֽ�ϵ�����ת��250 mL����ƿ������1.1������“��50%�ı�ͪ100 mL”������������
1.3 ��� ��ȡ��ϸ�ɷ�ĩ������4 g����50%��ͪ100 mLʹ�ܽ⣬����ֽ���ˣ���Һ��ȥ��ͪ�����䣬����������������ȡ2�Σ�ÿ��50 mL���ϲ���ȡҺ�����ɣ��ñ�ͪ5 mL�ܽ��������Ϊ����Ʒ��Һ��
2 ����Һ�Ʊ�
ȡ��������A��B��C������������Ʒ�ӱ�ͪ�Ƴ�ÿ1 mL��0.5 mg�Ļ����Һ����Ϊ����Ʒ��Һ��
3 ��������ú�4%�����Ƶ��ȼ���ά������Һ�Ʊ��Ĺ轺H����塣
4 ����������Ʒ��Һ�Ͷ���Ʒ��Һ������5 μL�������ӹ���Ʒ��Һ�ĵ�������ȷ���������Ƿ������ӡ�
5 չ�������ױ�-��������-��ͪ-�״���10:5:5:0.6��
6 չ����ʽ����15 ������չ��������չ��12 cm��ȡ�������ɡ�
7 ��ɫ; �ô�������Ѭ15 min����140 ����160 ������30 min��������ƣ�365 nm���¼��ӡ�
8 ɫ��ʶ��������Ʒɫ�������������Ʒɫ����Ӧ��λ���ϣ�����ͬ��ɫ��ӫ��ߵ㡣

|
1.����Gincosan
2.������
3.GIMKGO POWER
4.��������A��B��C��������
|
4. ��������A��B��C��������
5. ���������Ҷ��
|
6. �պ���δ�������Ҷ4μL
7. �պ��� 8μL
8. �պ��� 12μL
9. ��������A��B��C��������
|
�� ���
1 ����Һ�Ʊ�
1.1Ƭ������
1.1.1��ȡ��ϸ������2 g���Ӽ״�30 mL������2 h���˹�����Һ���ɣ������Ӽ״�1 mLʹ�ܽ⣬��Ϊ����Ʒ��Һ��
1.1.2��1.1.1�Ʊ��Ĺ���Ʒ��Һ������н϶����ʣ�Ӱ�챡��ɫ���룬�ɸ������������Ʊ�����ƷҺ����ȡ��ϸ������2 g�����Ҵ�40 mL�����Ȼ���30 min���˹�����Һ���ɣ�������0.3%����������Һ20 mL�ܽⲢת������Һ©���У���ϡ���ᣨ234→1000������pH 5��6��������������ҡ��ȡ2����ÿ��20 mL���ϲ���ȡҺ������ˮ��������ˮ���˹�����Һ���ɣ���������������2 mLʹ�ܽ⣬��Ϊ����Ʒ��Һ��
1.2 Һ������
ȡ����������20 mL���÷�Һ©���У�������������ҡ��ȡ���Σ�ÿ��20 mL���ϲ���ȡҺ������ˮ��������ˮ���˹�����Һ���ɣ���������������1 mLʹ�ܽ⣬��Ϊ����Ʒ��Һ��
2 ����Һ�Ʊ�
ȡ����ض���Ʒ���Ӽ״��Ƴ�ÿ1 mL��1 mg����Һ����Ϊ����Ʒ��Һ��
3 ���������0.5%�ȼ���ά����Ϊ�ϼ��Ĺ轺G����塣
4 ����������״����������Һ5 μL��10 μL������Һ5 μL����������и���غ����ϵͿ����ӵ�������
5 չ���������ȼ���-�״�-ˮ��28:7:1��
6 չ����ʽ������չ��12cm��ȡ�������ɡ�
7 ��ɫ;�ð�������Ѭ15 min��������ƣ�365 nm���¼��ӡ�
 |
8 ɫ��ʶ��������Ʒɫ�������������Ʒɫ����Ӧ��λ����������ͬ��ɫ��ӫ�����ߡ�
1. ϲ��Ѫ����������
2. �����
�� «��
1 ����Һ�Ʊ�
������������ ȡ��Ʒ��ĩ2 g���Ӽ״�30 mL��ˮԡ�ϼ������У���ҡ�����ӣ��˹�����Һ��Ϊ����Ʒ��Һ��
2 ����Һ�Ʊ�
ȡ«���ն���Ʒ���Ӽ״��Ƴ�ÿ1 mL��5 mg����Һ����Ϊ����Ʒ��Һ��
3 ��������轺G�������
4 ����������Ʒ��Һ�����Ʒ��Һ�ֱ����1 μL��2 μL��
5 չ��������������-�״�-ˮ��100��17��13����
6 չ����ʽ������չ��10 cm��
7 ��ɫ; ����10%�������ؼ״���Һ�����������365 nm���¼��ӡ�
8 ɫ��ʶ��������Ʒɫ���У��������Ʒɫ����Ӧ��λ����������ͬ��ɫ��ӫ��ߵ㡣
 |
1. ������«������1�Ž��� 1. «����
2. «���� 2. ʮ�˱��ư����ƽ���
ʮһ������ʳƷ����Ƥ����ɽ�������������صĺ����ⶨ
Determination of quercetin��keampferol��isorhamnetin in health food
1 ��Χ
�������涨�˱���ʳƷ����Ƥ�ء�ɽ���ء��������ص�Һ��ɫ�ײⶨ������
������������������Ҷ������Ҷ��ȡ��Ϊ��Ҫԭ�ϵı���ƷʳƷ����Ƥ�ء�ɽ���ء��������صIJⶨ��
2 ԭ��
��������ȡ��ˮ���ǰ����������Һ��ɫ���롢����������⣬�Ա���ʱ�䶨�ԣ�����귨������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1 �״�(CH3OH)��ɫ�״���
3.1.2 ����(H3PO4)��
3.1.3 ����(HCl)����������Լ37 %��
3.2 �Լ�����
3.2.1 ������Һ(1.5 mol/L)��ȡ����13.5 mL����ˮ����ʹ��100 mL��ҡ����
3.2.2 80%�״���Һ�����״���ˮ��80+20������Ȼ�Ͼ�����
3.3 ��Ʒ
3.3.1 ��Ƥ��(C15H10O7)��CAS����117-39-5������≥98%��
3.3.2 ɽ����(C15H10O6)��CAS���520-18-3������≥98%��
3.3.3 ��������(C16H12O7)��CAS���480-19-3������≥98%��
3.3 ����Һ������
3.4.1 ������Һ(1 mg/mL)���ֱ�ȷ��ȡ��Ƥ����ɽ�������������ر�Ʒ100 mg(ȷ��0.1 mg)����100 mL����ƿ�У��ü״��ܽⲢ�������̶���
3.4.2 ��Ƥ����ɽ�������������ػ�ϱ��м�Һ(200 μg/mL)���ֱ�ȷ��ȡ��Ƥ����ɽ�������������ر�����Һ��5.00 mL��25 mL����ƿ�У��������ඨ����
3.4.3 ��ϱ�ϵ�й���Һ���ֱ�ȷ��ȡ��Ƥ����ɽ�������������ػ�ϱ��м�Һ0.50 mL��1.00 mL��2.00 mL��3.00 mL��5.00 mL��10 mL����ƿ�У��������ඨ�����̶ȣ����Ƴ�����Ũ�ȷֱ�Ϊ10 μg/mL��20 μg/mL��40 μg/mL��60 μg/mL��100 μg/mL �Ļ�ϱ�ϵ�й���Һ��
4 �����豸
4.1 ��ЧҺ��ɫ�����������������л�����������UV����
4.2 ������ƽ������Ϊ 1 mg �� 0.1 mg��
4.3 ��������ϴ��������≥250W��
4.4 ˮԡ����
5 ��������
5.1 �����Ʊ�
5.1.1 ��Ƥ����ɽ���������������IJⶨ
ȡ�����������൱�ں���Ƥ�ء�ɽ���ء�������������Լ3 mg����ȷ��0.001 g������20 mL�״���������ȡ30 min���˹��������ü״�Լ5 mLϴ�ӣ�ϴҺ������Һ������20 mL������Һ(1.5 mol/L)��ˮԡ����ˮ��3 h����ȴ��ת����50 mL����ƿ�У��ü״��������̶ȣ���������0.45 μm����Ĥ���ˣ�ȡ����Һ����Ϊ��������Һ��
5.1.2 ������Ƥ����ɽ���������������IJⶨ
ȡ�����������൱�ں���Ƥ�ء�ɽ���ء�������������Լ3 mg����ȷ��0.001 g�����þ�����ƿ�������ܼ���80%�״���Һ20 mL���������ƶ�������������ȡ������250W��Ƶ��33kHz��20 min��ȡ�������䣬�ٳƶ���������80%�״���Һ�����ʧ��������ҡ�ȣ��˹���ȡ����Һ����Ϊ��������Һ��
5.2ɫ�ײο�����
5.2.1 ɫ������ C18 ����5 μm��100 Å��3.9 mm ID×150 mm��ͬ������ɫ������
5.2.2 ���£����¡�
5.2.3 ��Ⲩ����360 nm��
5.2.4 ���������״�+0.4%������Һ=50+50��
5.2.5 ���٣�1.0 mL/min��
5.2.6 ��������10 μL��
5.3 �����ߵ�����
��10 μL�Ļ�ϱ�ϵ�й���Һ�ֱ�ע��Һ��ɫ�����У������Ӧ�ķ�������Ա�����Һ��Ũ��(μg/mL)Ϊ�����꣬�Է����Ϊ�����꣬���Ʊ����ߡ�
5.4 ������Һ�IJⶨ
��10 μL ��������Һ(5.1.1)ע��Һ��ɫ�����У��Ա���ʱ�䶨�ԣ���÷���������ݱ����ߵõ�����Һ����Ƥ����ɽ����������������Ũ��(μg/mL)����10 μL ��������Һ(5.1.2)ע��Һ��ɫ�����У��Ա���ʱ�䶨�ԣ���÷���������ݱ����ߵõ�����Һ��������Ƥ����ɽ����������������Ũ��(μg/mL)��
6 ��������ı���
6.1 ��������Ƥ����ɽ�������������صĺ����ֱ�ʽ(1)������

ʽ����
Xi——��������Ƥ�ػ�ɽ���ػ��������ص���������λΪ��ÿ�ٿ�(g/100g)��
Ci——���ⶨ��Һ����Ƥ�ػ�ɽ���ػ�����������Ũ�ȣ���λΪ��ÿ����(μg/mL)��
V——���ⶨ��Һ�����ն����������λΪ����( mL)��
m——�����ij�����������λΪ��( g)��
100——��λת����
1000——��λת����
6.2 ��������Ƥ����ɽ�������������ص��ܺ�����ʽ(2)������

ʽ����
X——��������Ƥ����ɽ�������������ص�����������λΪ��ÿ�ٿ�(g/100g)��
X1——��������Ƥ�ص���������λΪ��ÿ�ٿ�(g/100g)��
X2——������ɽ���ص���������λΪ��ÿ�ٿ�(g/100g)��
X3——�������������ص���������λΪ��ÿ�ٿ�(g/100g)��
6.3 ����������Ҷ�ܻ�ͪ���յĺ�����ʽ(3)������

ʽ����
Y——����������Ҷ�ܻ�ͪ��������������λΪ��ÿ�ٿ�(g/100g)��
X——��������Ƥ����ɽ�������������ص�����������λΪ��ÿ�ٿ�(g/100g)��
2.51——����������
6.4 ������������Ƥ����ɽ�������������صĺ����ֱ�ʽ(4)������

ʽ����
Xi——������������Ƥ�ػ�����ɽ���ػ������������ص���������λΪ����ÿ��(mg/g)��
Ci——���ⶨ��Һ��������Ƥ�ػ�����ɽ���ػ���������������Ũ�ȣ���λΪ��ÿ����(μg/mL)��
V——���ⶨ��Һ�����ն����������λΪ����( mL)��
m——�����ij�����������λΪ��( g)��
1000——��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
8 ����
��������Ϊ 0.5 g���������Ϊ 50 mLʱ������������ֱ�Ϊ����Ƥ��0.02 mg/g��ɽ����0.03 mg/g����������0.05 mg/g��
��¼A
����Һ��������Һ����Һ��ɫ��ͼ
A.1��Ƥ����ɽ������������������Һɫ��ͼ����ͼA.1��

ͼA.1 ��Ƥ�ء�ɽ���ء��������ر���Һɫ��ͼ
A.2����Ƥ����ɽ�������������ص�������Һɫ��ͼ����ͼA.2��

ͼA.2 ����Ƥ�ء�ɽ���ء��������ص�������Һɫ��ͼ
ʮ��������ʳƷ�в谱��IJⶨ
Determination of theanine in health food
1 ��Χ
�������涨�˱���ʳƷ�в谱��ĸ�ЧҺ��ɫ�ײⶨ������
�����������ں�衢�̲��Ϊ��Ҫԭ�ϵı���ʳƷ�в谱�Ậ���IJⶨ��
2 ����ԭ��
. �������еIJ谱����ˮ��ȡ��ʹ�õȶ�ϴ������ЧҺ��ɫ���룬����������⣬�����������������귨��������
3 �Լ�
ע��������˵���������Լ���Ϊ��������ʵ����ˮ����GB/T 6682�涨һ��ˮҪ��
3.1 ��������(CF3COOH)��ɫ�״���
3.2 �谱���Ʒ(C7H14N2O3)��CAS��3081-61-6������≥98����
3.3 ��������ˮ��Һ��pH 3.0����ȡˮ���������ᣨ3.1��������pH 3.0��
3.4 �谱�������Һ��3.5 mg/mL������ȡ35.0 mg�谱����Ʒ����ȷ��0.1 mg������ˮ�ܽ������10 mL����ƿ����ˮ�������̶���
3.5 �谱���ʹ��Һ���ֱ�ȷ��ȡ�谱�������Һ��3.4��0.2 mL��0.4 mL��0.6 mL��0.8 mL��1.0 mL������10 mL����ƿ����ˮ�������̶����õ�Ũ�ȷֱ�Ϊ0.07 mg/mL��0.14 mg/mL��0.21 mg/mL��0.28 mg/mL��0.35 mg/mL�谱���ʹ��Һ��
4 �����豸
4.1 ��ЧҺ��ɫ���ǣ�����������������������м������
4.2 ����ˮԡ����
4.3 ���Ļ���4000 r/min��
4.4 ������ƽ������1 mg��0.1 mg��
4.5 pH�ơ�
5 ��������
5.1 ���������Ĵ�������ȡ����������������ȷ��0.001 g���൱�ں��谱��10 mg������ˮ30 mL����80 ��������ˮԡ���ϼ���40 min����ȴ�����ģ����˺�ת����50 mL����ƿ����ˮ�������̶ȣ����ȡ�������Һ��0.45 μmˮĤ��������
5.2 Һ�������Ĵ�����ȡһ������������ˮԡ�������ɣ�������ˮ�ܽ⣬ת����10 mL����ƿ����ˮ�������̶ȣ����ȡ���Һ��0.45 μmˮĤ��������
5.3 ɫ�ײο�����
5.3.1 ɫ������C18��,5 μm,4.6×250 mm��ͬЧ����
5.3.2 ��Ⲩ����203 nm��
5.3.3 ����������������ˮ��Һ��3.3����
5.3.4 ���٣�1.0 mL/min��
5.3.5 ��������10.0 mL��
5.3.6 ���£�35 ����
5.4 ɫ����
5.4.1 �����ߵ��Ʊ����ֱ�ȡ��ʹ��Һ��3.5��Ũ���ֱ�Ϊ0.07 mg/mL��0.14 mg/mL��0.21 mg/mL��0.28 mg/mL��0.35 mg/mL��10 μL����HPLC�������Ա���Ũ�ȶԷ�������Ʋ谱��ı��ع����ߡ�
5.4.2 �����ⶨ����������Һע����ЧҺ��ɫ�������Ա���ʱ��������-�ɼ����չ������ԣ����������������ɫ��ķ�����ɴӱ������������Ӧ�IJ谱���Ũ����
6�������
6.1�������谱���ĺ���
X= 

ʽ�У�
X—������Һ�в谱��ĺ�����g/100g��
C—������Һ�в谱���Ũ����mg/mL��
V—�������������mL��
m—����������g��
6.2�����ʾ���������������λ��Ч������
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
8 ����
����������ͼ������10 ng��
�� ¼ A
�������Ը�¼��
�谱�����ƷҺ��ɫ��ͼ
0.28 mg/mL�谱�����ҺҺ��ɫ��ͼ��ͼA.1��

ͼA.1 0.28 mg/mL�谱�����ҺҺ��ɫ��ͼ
ʮ��������ʳƷ����ζ�Ӵ��ס���ζ�Ӽ��غ����صIJⶨ
Determination of schisandrin��deoxyschisandrin��schisandrin B in health food
1 ��Χ
����������������ζ��Ϊ��Ҫԭ�������ı���ʳƷ����ζ�Ӵ��ס���ζ�Ӽ��غ����غ�������ЧҺ��ɫ���ⶨ��
2 ԭ��
�������е�ľ֬����ȡ��ʹ�õȶ�ϴ�ѷ����ЧҺ��ɫ���з��룬���������м������⣬����ɫ��ı���ʱ�����������ͼ���ԣ���귨�������������Ա���ζ��Ϊ��Ҫԭ�������ı���ʳƷ����ζ�Ӵ��ס���ζ�Ӽ��غ���������������
3 �Լ�
ע��������˵���������Լ���Ϊ��������ʵ����ˮ����GB/T 6682-2008һ��ˮҪ��
3.1 �״���CH3OH����ɫ�״���
3.2 ��ζ�Ӵ��ס���ζ�Ӽ��غ����ر�Ʒ������������98����
3.3 ��ζ�Ӵ��ס���ζ�Ӽ��غ����ر���Һ�����ƣ�������ζ�Ӵ��ס���ζ�Ӽ��غ����ر�����Һ��Ũ�ȷֱ�Ϊ2mg/mL�����Դ˴���Һ���Ƴɻ�ϱ�ϵ����Һ��Ũ�ȷ�ΧΪ0.02mg/mL��1mg/mL�����б���Һ���ü״����ơ�
4 ����
4.1 ��ЧҺ��ɫ���ǣ����������������������������
4.2 ��������ϴ����
4.3 ������ƽ������1mg��0.1mg��
5 ��������
5.1 ������������ȡ�������Ʒ�������൱�ں���ζ������30mg����ȷ��0.001g������20mL����ƿ�У�����״�Լ18mL��������ȡ20min��ȡ�������ô��䣬�Ӽ״����̶ȡ�������Һ��0.45μm�л�ϵ��Ĥ����Һ����ɫ������
5.2 �ⶨ
5.2.1 Һ��ɫ�ײο�����
5.2.1.1 ɫ������C18����5μm��100Å��4.6×250mm��ͬ������ɫ������
5.2.1.2 ��Ⲩ����254nm��
5.2.1.3 �ȶ���ϴ�������״�-ˮ=77:23��v/v����
5.2.1.4 ���٣�1mL/min��
5.2.1.5 ���£�35����
5.2.2 ɫ����
5.2.2.1 �����ߵ��Ʊ����������ϵ����Һ��ȡ10μL ��HPLC �������÷������Ũ�ȼ�����ζ�Ӵ��ס���ζ�Ӽ��غ����صı��ع����ߡ�
5.3.2.2 �����ⶨ��ȡ10μL ��������Һ���и�ЧҺ��ɫ�������Ծ��Ա���ʱ�����������ͼ���ԣ��÷����ͨ����ζ�Ӵ��ס���ζ�Ӽ��غ����صı����߶������������еĺ�����
6��������ı���
��������ζ�Ӵ��ס���ζ�Ӽ��غ����صĺ�����ʽ��1�����㣺
………………(1)
ʽ�У�X——��������ζ�Ӵ��ס���ζ�Ӽ��غ����صĺ�����mg/100g��
C —— ������Һ����ζ�Ӵ��ס���ζ�Ӽ��غ����صĺ�����mg/mL��
m —— ����������g��
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
8 ����
�������ļ���ֱ�Ϊ��ζ�Ӵ���0.02μg����ζ�Ӽ��� 0.03μg����ζ������0.02μg��
��������������Է�ΧΪ0.2��10μg��
ʮ��������ʳƷ�����յIJⶨ
Determination of adenosine in health food
1 ��Χ
�������涨�˱���ʳƷ�����յĸ�ЧҺ��ɫ�ײⶨ������
�����������ڱ���ʳƷ�����յIJⶨ��
2 ԭ��
������ˮ������ȡ���ø�ЧҺ��ɫ���вⶨ���Ա���ʱ�䶨�ԣ��������귨������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1 ��������(KH2PO4)��
3.1.2 �״�(CH3OH)��ɫ�״���
3.1.3 ������(SiO2)����ѧ����
3.2 �Լ�����
0.01mol/L����������Һ����ȡ1.36 g�������أ���ˮ�ܽⲢϡ����1000 mL����0.45 μm����Ĥ���˺�ʹ�á�
3.3 ��Ʒ
����(C10H13N5O4)������≥98%��������֤�����������֤��ı����ʡ�
3.4 ����Һ����
3.4.1 ���ձ�����Һ(1.0 mg/mL)�����ܳ�ȡ10 mg (��ȷ��0.1 mg)������Ʒ��10 mL ����ƿ�У���ˮ�ܽⲢ�������̶ȣ�ҡ�ȡ�
3.4.2 ���ձ��м�Һ(100 μg/mL)��������ȡ���ձ�����Һ2.5 mL��25 mL����ƿ�У���ˮϡ�����̶ȣ�ҡ�ȡ�
3.4.3 ���ձ�����Һ��������ȡ���ձ��м�Һ0.1 mL��0.2 mL��0.5 mL��1.0 mL��2.0 mL��5.0 mL��10 mL����ƿ�У���ˮϡ�����̶�����Ũ��Ϊ1.0 μg/mL��2.0 μg/mL��5.0 μg/mL��10.0 μg/mL��20.0 μg/mL��50.0 μg/mL�ı�����Һ��
4 �����豸
4.1 ������ƽ������Ϊ0.1 mg��0.01 mg��
4.2 ��������ȡ����
4.3 ���Ļ���
4.4 ��ЧҺ��ɫ���ǣ���������(UV)����������������(DAD)�������
5 ��������
5.1 �����Ʊ�
5.1.1 ��������
5.1.1.1 ��������
��ȡ���鲢��Ͼ��ȵ�����0.5 g~2 g (��ȷ��0.001 g) (���������Լ0.05 mg ~2.5 mg)��50 mL����ƿ�У�����ˮԼ30 mL��������ȡ20 min�����䣬��ˮϡ�����̶ȣ�ҡ�ȣ���3000 rpm/min����5 min����0.45μmˮϵ��Ĥ���˺�ȡ����Һ��Һ��ɫ�����á�
5.1.1.2 ����������
ȡ�������������������ﲢ���ȣ���ȡ0.5 g~2 g (��ȷ�� 0.001 g)�����������������������ɢ���ȣ���ȡ���в��� (��ȷ��0.001g�����������Լ0.05 mg ~2.5 mg)��ת����250 mL��������ƿ�У�����ȡ50 mLˮ����������ƿ�У����أ�����������ȡ20 min�����䣬��ˮ����������ҡ�ȣ����ó������3000r/min����5 min��ȡ����Һ��0.45 μmˮϵ��Ĥ���˺�����Һ��Һ��ɫ�����á�
5.1.1.3 Һ������
������ȡ���ȵ�����5.0 mL ~10.0 mL (���������Լ0.05 mg ~2.5 mg)��50 mL����ƿ�У�����ˮԼ30 mL��������ȡ20 min�����䣬��ˮϡ�����̶ȣ�ҡ�ȣ���3000r/min����5min����0.45μmˮϵ��Ĥ���˺�ȡ����Һ��Һ��ɫ�����á�
5.1.2 ϡ��
���������������ĺ�����ˮ�����ʵ���ϡ��(F)��ʹ������Һ������Ũ����1.0 μg/mL~50.0 μg/mL��Χ�ڡ�
5.2 Һ��ɫ���ο�����
5.2.1 ɫ������C18��4.6×250 mm��5 μm��ͬ������ɫ������
5.2.2 ���£����¡�
5.2.3 ��Ⲩ����254 nm��
5.2.4 �����ࣺ�״�+0.01mol/L����������Һ=10+90��
5.2.5 ���٣�1.0 mL/min��
5.2.6 ��������10 μL��
5.3 �����ߵ�����
������������Һ(3.4.3)�ӵ�Ũ�ȵ���Ũ�ȷֱ�ע���ЧҺ��ɫ�����У��ⶨ��Ӧ�ķ�������Ա�����Һ��Ũ��Ϊ�����꣬�Է����Ϊ�����꣬���Ʊ�����(����ҺҺ��ɫ��ͼ����¼A��ͼA.1)��
5.4 ������Һ�IJⶨ
����ͬɫ�������£���������Һ(5.1.1.1��5.1.1.2��5.1.1.3��5.1.2)ע���ЧҺ��ɫ�����У��Ա���ʱ�䶨�ԣ���÷���������ݱ������������Һ��������Ũ��(������ҺҺ��ɫ��ͼ����¼A��ͼA.2)��
6 ��������ı���
����������������ʽ(1)���㣺

ʽ��:
X——�����������ĺ�������̬������λΪ����ÿ�ٿ�(mg/100g)��Һ̬������λΪ����ÿ�ٺ���(mg/100 mL)��
ρ——���ݱ�������õ��Ĵ�����Һ��������Ũ�ȣ���λΪ��ÿ����(μg/mL)��
V——����ϡ�ͻ����������λΪ����(mL)��
F——ϡ�ͱ�����
m——����ȡ����������λΪ��(g)������ȡ���������λΪ����(mL)��
100——��λת����
1000——��λת����
���������ظ������»�õ��������ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ��10%��
8 ����
��������Ϊ2.0gʱ���������Ϊ50 mLʱ�����յĶ�����Ϊ1.5mg/100g��
��ȡ����Ϊ10 mLʱ���������Ϊ50 mLʱ�����յĶ�����Ϊ0.30mg/100mL��
��¼ A
����Һ��������Һ����ЧҺ��ɫ��ͼ
A.1 ���ձ���Һɫ��ͼ����ͼA.1��

ͼA.1 ���ձ���Һɫ��ͼ
A.2 �������յ�������Һɫ��ͼ����ͼA.2��

ͼA.2 �������յ�������Һɫ��ͼ
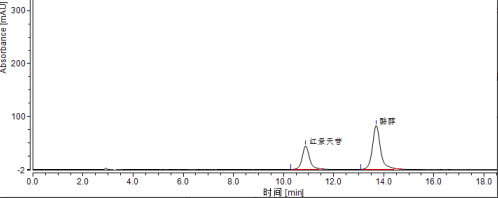
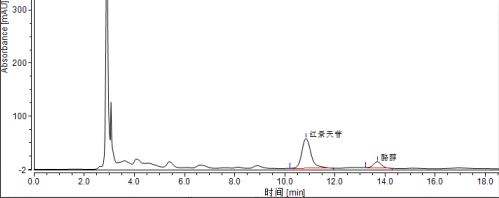
ʮ��������ʳƷ���������IJⶨ
Determination of total ginsenosides in health food
1 ��Χ
�����涨�˱���ʳƷ�������յķֹ��Ȳⶨ������
���������ڱ���ʳƷ�������պ����IJⶨ��
2 ԭ��
������ˮ��ȡ��������ɷ֣��������֬����ˮ������������ȡ���Ӻ������е�������ɷ��ڸ�����������������ȩ��Ӧ�������������Ϻ�ɫ�����÷ֹ��ȷ��ⶨ560 nm������������ȣ����ж�����
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T 6682 �涨��һ��ˮ��
3.4 �Լ�
3.1.1 Amberlite-XAD-2 �����֬����D-101�����֬����
3.1.2 �����������������ã�100-200Ŀ����
3.1.3 ������(CH3(CH2)2CH2OH)��
3.1.4 ��ˮ�Ҵ�(CH3CH2OH)��
3.1.5 �״�(CH3OH)��
3.1.6 ��ˮ(NH3)��
3.1.7 ������(HClO4)��
3.1.8 ������ (CH3COOH)��
3.1.9 ���ȩ(C8H8O3)��
3.2 �Լ�����
3.2.1 ���ȩ��Һ��5%������ȡ5g���ȩ���ӱ������ܽ���������100mL��������
3.2.2 ˮ������������Һ��ȡ��������������������ˮ�������ҡ������ʹ�ֲ㣬�ϲ�Һ�弴Ϊˮ������������
3.2.3 ����Һ��ȡ��ˮ40mL����ˮʹ��100mL�����ȡ�
3.3 ��Ʒ
�˲�����Re (C48H82O18)��
3.4 ����Һ������
�˲�����Re������Һ(0.2 mg/mL)��ȷ��ȡ10.0 mg �˲�����Re��Ʒ����ȷ��0.1 mg���� 50 mL����ƿ�У����״��ܽⲢ�������̶ȣ�ҡ�ȡ�
4 �����豸
4.1 ����/�ɼ��ֹ��ȼơ�
4.2 ��ƽ������Ϊ 1 mg �� 0.1 mg��
4.3 ��������ϴ����
4.4 ���Ļ���ת�� ≥4000 r/min��
4.5 ����ˮԡ����
5 ��������
5.1 �����Ʊ�
5.1.1 ��������
5.1.1.1 ��������
��ȡ�ѷ����Ͼ��ȵĴ�������1 g(��ȷ�� 0.001 g)�������������������������������������ƿ�У�����ˮ100.0 mL�����أ�����30 min�����䣬����ˮ�����ʧ������ҡ�ȣ����ã��˹�������Һ������
5.1.1.2Һ������
���Ҵ���Һ����������ȡ��Ͼ��ȵĴ�������10.0 mL���������������������������ˮԡ�ϻӾ��Ҵ�����ˮת����10mL����ƿ�У�����ˮϡ�����̶ȣ����ã����Ҵ����Һ��������ֱ��ȡ����
5.1.2 ȥ�����ʣ��ɸ�����Ʒ����ѡ������һ�ַ�����
5.1.2.1 �����֬��������
��10 mLע�����������ܣ���װ3cm�����֬���ϼ�1 cm����������������25 mL70%�Ҵ�ϴ������ȥϴ��Һ������Լ25 mLˮϴ������ζ����ȥϴ��Һ������1.0 mL�Ѵ����õ�������Һ��5.1.1.1��5.1.1.2������25 mLˮϴ������ȥϴ��Һ������25 mL������70%�Ҵ�ϴ���˲�������ϴ��Һ��ɫ���ռ�ϴ��Һ���������У�����60��ˮԡ�Ӹɣ������������״��ܽⲢת����10mL������ɫ���У����á�
5.1.2.2 ˮ������������ȡ��
ȡ5.1.1.1���±�����Һ25.0 mL�÷�Һ©���У���5.1.1.2���±�����Һ��ˮȫ��ת������Һ©���У����Ҵ���Һ������ֱ��ȡ10.0 mL������ˮ��Լ25mL������20 mLˮ������������3.2.2����ҡ��ȡ����ȡ������Һ����Ҫʱ�����ģ����ظ�����3�Σ��ϲ�������Һ��20 mL����Һ��3.2.3��ϴ�����ظ�����2������ȥ����Һ��������Һˮԡ���ɣ������ü״��ܽⲢת����25 mL��ƿ�У�Һ����Ʒ��ת����10 mL��ƿ�У����Ӽ״��������̶ȣ�ҡ�ȣ��˹���ȡ����Һ��������
5.2 �����ߵ�����
��ȡ�˲�����Re����Һ��3.4��0 mL��0.4 mL��0.6 mL��0.8 mL��1.0 mL��1.2 mL��10 mL������ɫ���У���ˮԡ�лӸ��ܼ�������0.2 mL���ȩ��������Һ��3.2.1�����ټ���0.8 mL�����ᣨ3.1.7�������ȣ�ʹ����ȫ���ܽ⣬��60��ˮԡ�м���10 min��ȡ������ԡ��ȴ����5.0 mL�����ᣨ3.1.8����ҡ�Ⱥ�������560 nm�������ⶨ����ȡ�
5.3 ������Һ�IJⶨ
ȡ5.1.2.1���±�����Һ��������ȡ5.1.2.2 ����������Һ1.0 mL��10 mL������ɫ���У���5.2 “��ˮԡ�лӸ��ܼ�……”�������Һͬ���ⶨ����ȡ�
5.4 ���У��������Ʒ�����ڱ������ţ�����У������5.1.2.1ͬ���Ʊ�������Һ��������ȡ5.1.2.2 ����������Һ1.0 mL��10 mL������ɫ��������ˮԡ�лӸ��ܼ�������0.2 mL��������3.1.8������5.2 “�پ��ܼ���0.8 mL��������3.1.7��……”��������ͬ���ⶨ����ȣ�����������У����
6 ��������ı���
���������������������˲�����Re�ƣ���ʽ��1������:
Ci ×V×100
Xi = ..........................................(1)
V0×m
ʽ��:
Xi——�������������ĺ��������˲�����Re�ƣ�����λΪ����ÿ�ٿ���mg/100g�������ÿ��������mg/100mL����
Ci——����������У�����ɱ�������ñ���Һ���˲�����Re��������λΪ����( mg)��
V——���ⶨ��Һ�Ķ����������λΪ����( mL)��
V0——���ⶨ��Һ����ɫ�������λΪ����( mL)��
m——�����ij�����������λΪ��( g)��
100——��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ��10% ��
8 ����
������������������Ϊ 1.0 g���������Ϊ 100 mLʱ�������գ����˲�����Re�ƣ��Ķ�����Ϊ0.5 mg/100g��Һ����������ȡ����Ϊ1.0����10.0��mL���������Ϊ1����10��mLʱ�������գ����˲�����Re�ƣ��Ķ�����Ϊ0.005 mg/100mL��
ʮ��������ʳƷ���ܻ�ͪ�IJⶨ
Determination of total flavonoid in health food
1 ��Χ
�������������Ժ���ͪ��ɷ�Ϊ��Ҫԭ�ϵı���ʳƷ���ܻ�ͪ�����IJⶨ��
2 ԭ��
������Ԥ�������Ӻ��Լ״���60%�Ҵ���Һ��ȡ��ͪ��ɷ֡������еĻ�ͪ��ɷֿɱ��������ƻ�ԭ������������������������������Һ���������¿��������� 2-�ǻ����ͪ��ʹ��Һ�������ijȺ�ɫ�����÷ֹ�ⷨ��510nm�������ⶨ����ȣ���«��Ϊ����Ʒ�����ñ����߷�������Ʒ���ܻ�ͪ�ĺ�����
3 �Լ�
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T 6682 �涨��һ��ˮ��
3.1 5������������Һ����ȡ5g�������ƣ�NaNO₂������ˮ�ܽ��100mL��
3.2 10����������Һ����ȡ��������Al(NO3)3•9H2O��10g����ˮ�ܽ��100mL��
3.3 ����������Һ��ȡ�������ƣ�NaOH��4.3g����ˮ�ܽ��100mL��
3.4 ʯ���ѣ�60��90�棩��
3.5 �Ҵ�(CH3CH2OH)��
3.6 �״�(CH3OH)��
3.7 «������Ʒ��Һ�� ȡ«������Ʒ���������ܳƶ����Ӽ״��ܽ⣬�Ƴ�Ũ��Ϊ0.2mg/mL�Ķ���Ʒ��Һ��
4������
4.1������/�ɼ��ֹ��ȼ���
4.2����������ȡ����
4.3�����Ļ���
4.4��������ƽ������1mg��0.1mg��
5����������
5.1 ��������������ȡ����������Һȡ������ɸ����������ܻ�ͪ�ĺ����ʵ��������Ա�֤�ⶨ�����ն�ֵ��0.3��0.7��Χ�ڣ�
5.1.1�����������������������ܳ�ȡ����������ĩ��������������0.4g��M������������ȡ���У���ʯ���ѣ�60��90�������Ȼ�����ȡ����ȡҺ��ɫ����ȥʯ����Һ��������ȥʯ���ѣ�ת����������ƿ�У����ܼӼ״�25mL��V1�����������ƶ���������������30���ӣ����������£��ƶ��������ü״������ʧ��������ҡ�ȣ����ģ�ȡ����Һ��Ϊ����Ʒ��Һ��
5.1.2��Һ��������������ȡ����Ʒ2mL��M������25mL��V1����ƿ�У���60%�Ҵ��ܽⲢϡ�����̶ȣ�ҡ�ȣ���Ϊ����Ʒ��Һ��
5.2��«������Ʒ�������Ʊ���������ȡ«������Ʒ��Һ��3.7�0.0��1.0��2.0��3.0��4.0��5.0��6.0mL���ֱ���25mL��V3����ƿ�У���ˮ��6mL������5%����������Һ1mL��ҡ�ȣ�����6min����10%��������Һ1mL��ҡ�ȷ���6min��������������Һ10mL��ҡ�ȣ��ټ�ˮ���̶ȣ�ҡ�ȣ�����15���ӣ���0.0mL����Ʒ��Һ�Ƶõ��ܼ�Ϊ�հף��ڲ���510nm���ֱ�ⶨ������ֵ���������Ϊ������(A)������ƷŨ��Ϊ�����꣨mg/mL�������Ʊ����ߡ�
5.3 ��Ʒ�ⶨ��������ȡ����Ʒ��Һ2mL��V2������25mL��ƿ������5.2������ˮ��6mL��……������510nm�������ⶨ����ȣ�ͬ���������ӱ������϶�������Ʒ��Һ�к��ܻ�ͪ��Ũ��(C)��������Ʒ���ܻ�ͪ�ĺ�����X����
6 ��������ı���
C×V1×V3×100
X = ————————
V2×M×1000
ʽ�У�
X—�������ܻ�ͪ�ٷֺ�������«��(C27H30O16)�ƣ� g/100g��mL����
C—�������϶�������Ʒ��Һ���ܻ�ͪ��Ũ�ȣ�mg/mL��
V1—�������������mL��
V2—��ȡ����Һ�����mL��
V3—��ɫ���������mL��
M—����ȡ������g(mL)��
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
ͬһ����������ƽ�в��Խ���ľ��Բ�ֵ���ó�������ƽ��ֵ�ģ� 15%��������Ʒ����10%��Һ����Ʒ����
ע����Ʒ����ɫʱ���ɲ�����Ʒ�����ӷ�����0�Źܵ��㣬���Ʊ����ߣ���������Ʒ��ɫ���š�
8 ����
�������ļ���ޣ�LOD��Ϊ50ng�������ޣ�LOQ��200ng��
��������������Է�Χ����«���ƣ���9.9��59.2������Է�Χ��
ʮ��������ʳƷ���Ǿ����������ȵIJⶨ
Determination of deacetylationg degree of chitosan in health food
1 ��Χ
�������涨�˱���ʳƷ�пǾ���������������λ�ζ��ⶨ������
2 ԭ��
�ù�����������Һ�ܽ����������������������������������еİ�����Ħ����Ϻ���Һ�к��й��������ᡣ������������Һ�ζ�ʱ���������������к��������ᣬ��ҺpH �����仯������һ��“ͻԾ”��Ȼ�������������к��������������а�����ϵ����ᣬ�ﵽ�ζ��ȵ��ʱ����ҺpH ���ֵڶ���“ͻԾ”��������“ͻԾ”֮�����ĵ���������������������еİ����������Ӷ��õ����������������ȡ�
3 �Լ��Ͳ���
3.1 ������ζ�Һ��c(HCl)=0.1 mol/L��
3.2�������Ʊ��ζ���Һ��c(NaOH)=0.1 mol/L��
4 �����豸
4.1 ��Ž�������
4.2 ��ȼƻ��λ�ζ��ǡ�
4.3 ��ƽ������1mg��
4.4 ���ȸ����䡣
5 ��������
5.1 ��������
��ȡ��105°C ± 2°C��������ص�����0.2 g����ȷ��0.0001 g������30 mL ������ζ���Һ����������ȫ�ܽ⣬�ټ�50 mL ˮ�����ȡ�
5.2 �ζ�
���������Ʊ��ζ���Һ�ζ�5.1�õ�����Һ������pH��ÿ�μ�0.5 mL ��¼һ�Ρ����μ���pH�ӽ�“ͻԾ”ʱ����εζ�����¼�������Ʊ��ζ���Һ���������Ӧ��pH���õ�pH-�������Ʊ��ζ���Һ������ߣ��ҳ���“ͻԾ”�㡣��¼����ͻԾ��������ĵ��������Ʊ��ζ���Һ�������ֵ��
6 ��������ı���
�������ȵ���������w����ʽ ( 1 ) ������

ʽ����
ΔV——����“ͻԾ”��֮�����ĵ��������Ʊ��ζ���Һ���������λΪ����( mL )��
c ——�������Ʊ��ζ���Һ��Ũ�ȣ���λΪĦ��ÿ��( mol/L )��
10-3——��λ����ϵ����
16——������Ħ����������λΪ��ÿĦ��( g/moL )��
m ——�����пǾ��ǵ���������λΪ��( g )��
0.0994——���۰�������( 16/161 )��
ʵ������ƽ�вⶨ���������ƽ��ֵΪ�����ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ��2%��
ʮ�ˡ�ø���ԵIJⶨ����
Determination of activity of lumbrokinase
1 ��Χ
�����涨��ø����ø���ԵIJⶨ������
2 ԭ��
����ø�۾���ֱ�Ӻͼ���ܽ�Ѫ��ά�������ã�������������֬��—��ά����ƽ�巨��ͨ��ø��Ʒ�õ����Է��̣�����Ʒ�������Է��̼�������ø��Ч�ۡ�
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1 ���������(Na2HPO4·12H2O)��
3.1.2 ���������(NaH2PO4·2H2O)��
3.1.3 �Ȼ���(NaCl)��
3.1.3 ����(HCl)��
3.1.4 ��ά����ԭ��
3.1.5 ��Ѫø��
3.1.6 ø����Ʒ��
3.1.7 ��֬�ǡ�
3.2 �Լ�����
3.2.1 0.01 mol/L�����λ���Һ��ȡ���������3.58 g����ˮʹ�ܽⲢϡ����100 mLΪAҺ��ȡ���������0.78 g����ˮʹ�ܽⲢϡ����500 mLΪBҺ����A��B��Һ�����pH7.8��
3.2.2 ������Һ��ȡ0.01 mol/L�����λ���Һ��pH7.8����0.9%�Ȼ�����Һ��1:17����ϡ�
3.2.3 1.5%��֬��Һ��ȡ��֬��1.5 g���ӹ�����Һ100 mL�ܽ⡣
3.2.4 ��ά����ԭҺ��ȡ��ά����ԭ�������ӹ�����Һ�Ƴ�ÿ1 mL�к�1.5 mg��ά����ԭ����Һ��
3.2.5 ��Ѫø��Һ��ȡ��Ѫø��������0.9%�Ȼ�����Һ�Ƴ�ÿ1 mL����1 BP��λ����Һ��
3.2.6 ��֬��-��ά����ԭƽ���Ʊ�: ȡ��ά����ԭ��Һ39 mL�����ձ��У��߽�����55 °C��֬����Һ39 mL����Ѫø��Һ3.0 mL���������ȣ����ٵ���ֱ��14 cm�������������У�����ˮƽ����1Сʱ����ף����á�
3.2.7 ��Ʒ��Һ�Ʊ�: ȡø��Ʒ����0.9%�Ȼ�����Һ�Ƴ�Ũ�ȷֱ�Ϊÿ1 mL�к���10000��8000��6000��4000��2000ø��λ����Һ��
3.2.8 ������Һ�Ʊ�: ȡ��Ʒ��������ϸ�����ܳƶ�����0.9%�Ȼ�����Һ����10 min�����ݡ�ҡ�ȡ�ȡ����Һ�����ݱ����߷�Χ�����ʵ�ϡ�͡�
4 �����豸
4.1 ���������䡣
4.2 �����С�ܣ�φ3 mm��
4.3 ��ƽ������1 mg��
4.4 pH�ơ�
5 ��������
������ȡø��Ʒ��Һ��������Һ��10 µL���ֱ����ͬһ��֬��-��ά����ԭƽ���ϲ�ͬ��С���У��Ӹǣ���37°C�������з�Ӧ18Сʱ��ȡ�����ÿ��߲����ܾ�Ȧ��ֱֱ����ȡƽ��ֵ������Ȧ���������ø��Ʒ��λ���Ķ���Ϊ�����꣬��Ȧ����Ķ���Ϊ�����꣬��������ع鷽�̣�������Ʒ��Ȧ����Ķ�����������ع鷽�̣����㹩��ƷЧ�۵�λ������Ʒ����ƷӦ��������ȡƽ��ֵ��
6 ��������ı���
�����й�ʽ����ø�Ļ��ԣ�
S=(D/2)2×3.14
lgS = a×lgU + b
����
S ——��Ȧ���������λΪmm2��
D——��Ȧ��ֱ������λΪmm��
U——ø�Ļ��ԣ���λΪU/mL��
a——ø��Ʒ����õ������Է���б�ʣ�
b——ø��Ʒ����õ������Է��̽ؾࡣ
ʮ�š�����ʳƷ���������IJⶨ
Determination of total anthraquinone in health food
1 ��Χ
�������涨�˱���ʳƷ���������ķֹ��Ȳⶨ������
�����������ڱ���ʳƷ���������IJⶨ��
2 ԭ��
��������ˮ����л��ܼ���ȡ�������������ǻ������������ڼ�����Һ���Ժ�-�Ϻ�ɫ��Ӧ��Borntrager��Ӧ�� �����÷ֹ��ȷ����Ա����߶�����⡣
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T 6682 �涨������ˮ��
3.5 �Լ�
3.1.1 ����(HCl)
3.1.2 ��ˮ(NH3·H2O)
3.1.3 ���ȼ���(C2H6Cl2)
3.1.4 �������ƣ�NaOH��
3.1.5 �״� (CH3OH)
3.2 �Լ�����
3.2.1 25%���ȡ����68 mL����ˮϡ����100 mL��
3.2.2 4%����Һ��ȡ��ˮ16 mL����ˮϡ����100 mL��
3.2.3 10%����������Һ����ȡ��������10g����ˮ�ܽⲢ������100 mL��
3.2.4 ��ϼ���Һ�������10%�������ƺ�4%����Һ��ϡ�
3.3 ��Ʒ
1,8-���ǻ�����������≥99.0%��
3.4 ����Һ������
1,8-���ǻ���������Һ�����ܳ�ȡ1,8-���ǻ�������Ʒ10mg����25mL����ƿ�У��Ӽ״��ܽⲢϡ�����̶ȣ�ҡ�ȣ��Ƴ�0.4mg/mL����Һ��
4 �����豸
4.1 �ֹ��ȼ�
4.2 ��ƽ:����Ϊ 1 mg �� 0.1 mg��
4.3 ����ˮԡ��
5 ��������
5.1 ������
�ֱ�����ȡ1,8-���ǻ���������Һ0mL��0.2mL��0.4mL��0.6mL��0.8mL��1.0mL��25mL����ƿ�У��ӻ�ϼ���Һ���̶ȣ����ȣ��ڰ�������30���ӡ��Ի�ϼ���ҺΪ�հף���525nm���������ֱ�ⶨ����ȡ���Ũ�ȣ�mg/mL��Ϊ�����꣬�����Ϊ�����꣬���Ʊ����ߡ�
5.2�����ⶨ
��ȡ�ѷ����Ͼ��ȵĴ�������0.1��0.5g���൱��ÿg��������2mg��17mg��,���ܳƶ�����100mLԲ����ƿ�У����ܼ���״�-���ᣨ10:1�������Һ25mL,���أ���80��ˮԡ�м��Ȼ���30���ӣ����䣬�ü״������ʧ��������ҡ�ȣ��˹���������ȡ����Һ15mL����Һ©���У���ˮ25mL,�ö��ȼ���160mL��ȡ4�Σ�50mL��50mL��30mL��30mL�����ϲ����ȼ���Һ������ˮϴ�������ԣ���ȥˮϴҺ�����ȼ����ת���������������ɣ��ü״��������ܽⲢת����10mL����ƿ�У��ü״��������̶ȣ�ҡ�ȣ���Ϊ������Һ��������ȡ������Һ2mL, ��25mL��ƿ�У��ӻ�ϼ���Һ���̶ȣ����ȣ��ð�������30���ӣ�ȡ����Ѹ����ȴ�����£����أ����ӻ�ϼ���Һ��ԭ��������ҡ�ȣ��ⶨ����ȡ����ݻع鷽�̼����������������ĺ�����
6 ��������ı���
������������������ʽ(1)����:
C×V×100
X = ..........................................(1)
m
ʽ��:
X——�������������ĺ�������λΪ����ÿ�ٿ�(mg/100g);
C——�ɱ����߲�òⶨ��������������Ũ�ȣ���λΪ����ÿ����( mg/mL);
V——���ⶨ���������ն����������λΪ����( mL);
m——�����ij�����������λΪ��( g);
100——��λת��;
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ��10%��
8 ����
�������ļ����Ϊ1.3μg��
��ʮ������ʳƷ��10-�ǻ���ϩ��IJⶨ
Determination of 10-hydroxy-2-decenoic acid in health food
1 ��Χ
�������涨�˱���ʳƷ��10-�ǻ���ϩ������ЧҺ��ɫ�ײⶨ������
�����������ڱ���ʳƷ��10-�ǻ���ϩ��IJⶨ��
2 ԭ��
�������Ҵ���״���ȡ�����ø�ЧҺ��ɫ���вⶨ���Ա���ʱ�䶨�ԣ��������귨������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1����(HCL)��
3.1.2 �״�(CH3OH)��ɫ�״���
3.1.3 ��ˮ�Ҵ�(C2H6O)��
3.2 �Լ�����
3.2.1 0.1mol/L�����ȡ����9mL������ע��1000mLˮ��
3.2.2 0.03mol/L�����ȡ0.1mol/L����300mL������700mLˮ��
3.2.3 0.01mol/L�����ȡ0.1mol/L����100mL������900mLˮ��
3.3 ��Ʒ
10-�ǻ�-2-��ϩ��(C10H18O3)������≥98%��������֤�����������֤��ı����ʡ�
3.4 ����Һ������
3.4.1 10-�ǻ���ϩ�������Һ��1.0mg/mL�������ܳ�ȡ10 mg (��ȷ��0.1 mg) 10-�ǻ�-2-��ϩ���Ʒ��10 mL ����ƿ�У�����ˮ�Ҵ��ܽⲢ�������̶ȣ�ҡ�ȡ�
3.4.2 10-�ǻ���ϩ�������Һ���ֱ�ȷ��ȡ10-�ǻ���ϩ�������Һ0.02 mL��0.1 mL��0.2 mL��0.5 mL��1.0 mL �� 10.0 mL����ƿ�У�����ˮ�Ҵ�ϡ�����̶ȣ���Ũ��Ϊ2.0 μg/mL��10.0 μg/mL��20.0 μg/mL��50.0 μg/mL��100.0μg/mL�ı�����Һ��
4 �����豸
4.1 ������ƽ������0.1mg��0.01mg��
4.2 ��������ȡ����
4.3 ���Ļ���
4.4 ��ЧҺ��ɫ���ǣ���������(UV)����������������(DAD)�������
5 ��������
5.1 �����Ʊ�
5.1.1 ��������
5.1.1.1 ��������
��ȡ���鲢��Ͼ��ȵ�����0.5 g~2 g (��ȷ��0.001 g) (���������Լ0.1 mg ~5 mg)��50 mL����ƿ�У���0.01mol/L����2 mL������ˮ�Ҵ�30 mL��������ȡ20 min�����䣬����ˮ�Ҵ�ϡ�����̶ȣ�ҡ�ȣ���3000r/min������10 min����0.45μm�л�ϵ��Ĥ���˺�ȡ����Һ��Һ��ɫ�����á�
5.1.1.2 ����������
ȡ�������������������ﲢ���ȣ���ȡ0.5 g~2 g (��ȷ�� 0.001 g)�� ���������������������ɢ���ȣ���ȡ���в���(��ȷ��0.001g�����������Լ0.1 mg ~5 mg)��ת����250 mL��������ƿ�У�����ȡ0.01mol/L����2 mL����ˮ�Ҵ�48 mL����������ƿ�У����أ�����������ȡ20 min�����䣬����ˮ�Ҵ�����������ҡ�ȣ����ó����ȡ���ֻ���Һ3000r/min����10 min����0.45μm�л�ϵ��Ĥ���˺�ȡ����Һ��Һ��ɫ�����á�
5.1.1.3 Һ������
������ȡ���ȵ�����5.0 mL ~10.0 mL (���������Լ0.1 mg ~5 mg)��50 mL����ƿ�У��Ӽ״�30 mL��������ȡ20 min�����䣬���״�ϡ�����̶ȣ�ҡ�ȣ���3000r/min������10 min����0.45μm�л�ϵ��Ĥ���˺�ȡ����Һ��Һ��ɫ�����á�
5.1.2 ϡ��
����������10-�ǻ���ϩ��ĺ�������Ӧ�ܼ������ʵ���ϡ��(F)��ʹ������Һ��10-�ǻ���ϩ��Ũ����2.0 μg/mL~100.0 μg/mL��Χ�ڡ�
5.2 Һ��ɫ�ײο�����
5.2.1 ɫ������C18��4.6×250mm��5μm��ͬ������ɫ������
5.2.2 ���£�35�档
5.2.3 ��Ⲩ����210nm��
5.2.4 �����ࣺ�״�+0.03mol/L����+ˮ=55+10+35��
5.2.5 ���٣�1.0mL/min��
5.2.6 ��������10μL��
5.3 �����ߵ�����
��10-�ǻ���ϩ��������Һ(3.4.2)�ӵ�Ũ�ȵ���Ũ�ȷֱ�ע���ЧҺ��ɫ�����У��ⶨ��Ӧ�ķ�������Ա�����Һ��Ũ��Ϊ�����꣬�Է����Ϊ�����꣬���Ʊ�����(����ҺҺ��ɫ��ͼ����¼A��ͼA.1)��
5.4 ������Һ�IJⶨ
����ͬɫ�������£���������Һ(5.1.1.1��5.1.1.2��5.1.1.3��5.1.2)ע���ЧҺ��ɫ�����У��Ա���ʱ�䶨�ԣ���÷���������ݱ������������Һ��10-�ǻ���ϩ����Ũ��(������ҺҺ��ɫ��ͼ����¼A��ͼA.2)��
6 ��������ı���
������10-�ǻ���ϩ��������ʽ(1)���㣺

ʽ�У�
X——������10-�ǻ���ϩ���ĺ�������̬������λΪ��ÿ�ٿ�(g/100g)��Һ̬����Ϊ��ÿ�ٺ���(g/100mL)��
ρ——���ݱ�������õ��Ĵ�����Һ��10-�ǻ���ϩ����Ũ�ȣ���λΪ��ÿ����(μg/mL)��
V——����ϡ�ͻ����������λΪ����(mL)��
m——����ȡ����������λΪ�� (g)������ȡ���������λΪ����(mL)��
F——ϡ�ͱ�����
100——��λ���㣻
1000——��λ���㡣
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ��5%��
8 ����
��������Ϊ2.0 gʱ���������Ϊ50 mLʱ��10-�ǻ���ϩ���Ķ�����Ϊ0.5 mg/100g��
��ȡ����Ϊ10.0 mLʱ���������Ϊ50 mLʱ��10-�ǻ���ϩ���Ķ�����Ϊ0.1 mg/100mL��
��¼ A
����Һ��������Һ������ЧҺ��ɫ��ͼ
A.1 10-�ǻ�-2-��ϩ�����Һɫ��ͼ����ͼA.1��

ͼA.1 10-�ǻ�-2-��ϩ������Һɫ��ͼ
A.2 ����10-�ǻ�-2-��ϩ��Ĵ�����Һɫ��ͼ����ͼA.2��

ͼA.2 ����10-�ǻ�-2-��ϩ����������Һɫ��ͼ
��ʮһ������ʳƷ���ʹ�������XL IX�IJⶨ
Determination of gypenoside XL IX in health food
1 ��Χ
�����涨�˱���ʳƷ���ʹ�������XL IX����ЧҺ��ɫ�ײⶨ������
�����������Խʹ�������ӹ�ƷΪ��Ҫԭ�ϵı���ʳƷ�нʹ�������XL IX�����IJⶨ��
2 ԭ��
���������״���ȡ���ø�ЧҺ��ɫ���ⶨ�����ݱ���ʱ�䶨�ԣ���귨������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊɫ������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1 �״���CH3OH����
3.1.2 ���棨CH3CN����
3.2 ��Ʒ
�ʹ�������XL IX (C52H86O21��CAS�ţ�94987-08-3)������≥99.0%��
3.3 ����Һ������
3.3.1 �ʹ�������XL IX������Һ(5 mg/mL)����ȡ��60 ± 5 °C��40 kpa~53 kpa�����¼�ѹ����3 h�Ľʹ�������XL IX������25 mg (��ȷ��0.01 mg)�� 5 mL ����ƿ�У��ü״��ܽⲢ�������̶ȣ�ҡ�ȡ�����ҺӦ������-18°C �����С�
3.3.2 �ʹ�������XL IX������Һ����ȡ�ʹ�������XL IX������Һ��������Ũ�ȷֱ�Ϊ0.005 mg/mL��0.01 mg/mL��0.02 mg/mL��0.1 mg/mL��0.5 mg/mL��1 mg/mL ��2 mg/mL�ı�����Һ������ʱ���ơ�
4 �����豸
4.1 ��ЧҺ��ɫ���ǣ����������������м������
4.2 ��ƽ������Ϊ0.1 mg �� 0.01 mg��
4.3 ��������ϴ��������Ϊ800W��
4.4 ����Ĥ��0.45 μm���л��ࡣ
5 ��������
5.1 �����Ʊ�
5.1.1 ��������
ȡ20������Ƭ�������������з��顢���ȣ���ȡ���̬�������ȣ������ҳ�ȡ�������ȷ��ȡ��������0. 5 g (��ȷ��0.0001 g) �þ����Թ��У����ܼ���״�10 mL����������������5 min����ȴ�����£�ҡ�ȣ�����Ĥ(0.45 μm���л���)��ȡ��Һ�����⡣
5.1.2 Һ̬����
��ȡҡ�ȵ�����0.5 mL��10 mL����ƿ�У�����״�8 mL����������5 min����ȴ�����£��ü״�������10 mL������Ĥ(0.45 μm���л���)��ȡ��Һ�����⡣
5.2 ɫ�ײο�����
5.2.1 ɫ������ʮ�����������Ϲ轺��������100 mm���ھ�4.6 mm������3 μm���������൱�ߡ�
5.2.2 �����ࣺ������Ϊ������A����ˮΪ������B������1 �еĹ涨�����ݶ�ϴ�ѡ�
��1 �������ݶ�ϴ�ѱ�
|
ʱ��(min)
|
����(mL/min)
|
������A(%)
|
������B(%)
|
|
0
|
0.5
|
25
|
75
|
|
15
|
0.5
|
35
|
65
|
|
35
|
0.5
|
45
|
55
|
|
40
|
0.5
|
45
|
55
|
|
41
|
0.5
|
25
|
75
|
5.2.3 ��Ⲩ����203 nm ��
5.2.4 ���£�40 °C��
5.3 ϵͳ������
�������������ʹ�������XL IX�����Ӧ������10000��
5.4 �����ߵ�����
��10 μL�ı�ϵ�и�Ũ����Һ(3.3.2) ���Է����Ϊ�����꣬�Ա�����Һ�нʹ�������XL IX��Ũ��Ϊ�����꣬���Ʊ����ߡ�(����ҺҺ��ɫ��ͼ����¼A��ͼA.1 )��
5.5 ������Һ�IJⶨ
��10 μL������Һע���ЧҺ��ɫ���ǣ��Ա���ʱ�䶨�ԣ�ͬʱ��¼����������ݱ����ߵõ�����Һ�нʹ�������XL IX��Ũ��(������ҺҺ��ɫ��ͼ����¼A��ͼA.2)��
6 ��������ı���
�����ʹ�������XL IX������ʽ(1)������

ʽ����
X ——�����нʹ�������XL IX�ĺ�������λΪ��ÿ�ٿ�(g/100g)���ÿ����(g/100mL)��
ρ —— ��Һ�нʹ�������XL IX��Ũ�ȣ���λΪ mg/mL��
V—— ϡ���������λΪ����(mL)��
m——�������������������λΪ��( g )�����( mL );
100—��λת����
1000—��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ��10% ��
8 ����
��������Ϊ0.5 g��0.5 mL���������Ϊ10 mLʱ���ʹ�������XL IX�ļ����Ϊ0.005 g/100 g��0.005 g /100 mL��������Ϊ0.010 g/100 g��0.010 g /100 mL��
��¼ A
����Һ��������ҺҺ��ɫ��ͼ
A.1 �ʹ�������XL IX����Һɫ��ͼ����ͼ A.1��

ͼ A.1 �ʹ�������XL IX����Һɫ��ͼ
A.2 ���нʹ�������XL IX��������Һɫ��ͼ����ͼ A.2��

ͼ A.2 �����ʹ�������XL IX��������Һɫ��ͼ
��ʮ��������ʳƷ�������������IJⶨ
Determination of Glucosamine in health food
1 ��Χ
�������涨��������������Ϊ��Ч�ɷֵ�Ӳ��������������Ƭ����װ�ı���ʳƷ�������������IJⶨ������
�����������ڱ���ʳƷ�������������ĺ����ⶨ��
��������һ������ֹ��ȷ��ļ����Ϊ1.5μg���ڶ�����ЧҺ��ɫ��-�����ⷨ�ļ��Ũ��Ϊ4μg /mL��
��һ�� ����ֹ��ȷ�
2 ԭ��
��Ʒ��D-������������������ˮ�ܽ�����������ͪ���Զ��װ�������ȩ��Һ��ɫ֮�������Լ��հ���ҺΪ�αȣ���D-���ᰱ��������Ϊ����Ʒ����525 nm��������ɫ�ⶨ��
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1������ͪ(C5H8O2)��
3.1.2��ȩ�Ҵ�(C2H5OH)��
3.1.3̼����(NaCO3)��
3.1.4�Զ��װ�������ȩ(C9H11NO)��
3.1.5����(HCl)��
3.1.6ʯ���ѣ��г� 30°C~60°C��
3.2 �Լ�����
3.2.1 ������ͪ��Һ��ȡ������ͪ2 mL����0.5 mol/L̼������Һ��50 mL������ǰ���ơ�
3.2.2 �Զ��װ�������ȩ��Һ���Զ��װ�������ȩ0.8 g������ȩ�Ҵ�15 mL������15 mL��ҡ�ȣ�����ǰ���ơ�
3.3 ��Ʒ
D-���ᰱ��������������≥99.0%��
3.4 ����Һ������
3.4.1 ���ᰱ��������������Һ(1.0 mg/mL)����ȷ����105�����4Сʱ�����ص����ᰱ�������Ƕ���Ʒ10mg������10mL����ƿ�У���ˮ�������̶ȣ�ҡ�ȣ����á�
3.4.2���ᰱ������������Һ(0.1 mg/mL)��������ȡ1.0 ml������Һ����ˮ������10ml����Ϊ����Һ��
4 �����豸
4.1 ����ֹ��ȼ�������525±2nm��
4.2 ��ƽ������Ϊ0.1 mg��
4.3 �����䡣
4.4 ����ˮԡ����
4.5 ���Ļ���ת�� ≥3000 r/min��
4.6 ��������ϴ����
5 ��������
5.1 �����Ʊ�
5.1.1Ƭ����Ӳ��������
��ȡ�ѷ����Ͼ��ȵĴ���������Ӳ���ҳ�ȡ��Ͼ��ȵ���������Լ0.1~0.5g(��ȷ�� 0.0001 g)����100mL��ƿ�У���ˮ�ó�����ʹ���ܽ⣬��ˮϡ�����̶ȣ�ҡ�ȣ��˹���������ȡ����Һ��������50mL��ƿ�У���ˮϡ�����̶ȣ�ҡ�ȣ����ù���Ʒ��Һ��
5.1.2 ��������Ʒ
��ȡ��Ͼ��ȵ�������Ʒ 0.1 g~0.5g (��ȷ�� 0.0001 g) ��50 mL���Ĺ��У�����50mLʯ��������ҡ�ܽ⣬3000 r/min ����10 min������ȥ�ϲ���Һ�����õ������ɣ���ˮ������ȫ��ת����100mL��ƿ�У�����ˮ�ִ�ϴ�ӣ��ϲ���Һ����ƿ�У�������ʹ���ܽ⣬��ˮϡ�����̶ȣ�ҡ�ȣ��˹���������ȡ����Һ��������50mL��ƿ�У���ˮϡ�����̶ȣ�ҡ�ȣ����ù���Ʒ��Һ��
5.2 ������������
��ȡ���ᰱ������������Һ0��0.2��0.4 ��0.6��0.8��1.0��1.2mL����Ʒ��Һ����25mL������ɫ���У���ˮ��5mL���ֱ���������ͪ��Һ��ȡ������ͪ2 mL����0.5 mol/L̼������Һ��50 mL������ǰ���ƣ�1 mL��ҡ�ȣ��÷�ˮԡ�м���25���ӣ�ȡ�����ñ�ˮѸ����ȴ����ȩ�Ҵ�3.0 mL��60��ˮԡ�б���10���ӣ��ټӶԶ��װ�������ȩ��Һ1.0 mL��ǿ����ҡ��������60��ˮԡ�б���1Сʱ����ȴ�����¡��÷ֹ��ȼ���525 nm���������Լ��հ���ҺΪ�αȣ�1cm��ɫ��ⶨ�����ֵ����D-���ᰱ��������������μg��Ϊ�����꣬�����ֵΪ�����꣬���Ʊ����ߡ�
5.3 ������Һ�IJⶨ
������ȡ����Ʒ��Һ5.0mL���ֱ���25mL������ɫ���У���������ͪ��Һ1 mL�����°�“������”���в������ⶨ����Ʒ��Һ�����ֵ���ӱ������˵����������Ǻ��������㣬���á�
6 ��������ı���
���������������ǣ������ᰱ�������Ǽƣ�������ʽ(1)����:
Ci ×V×F×100
Xi = ..........................................(1)
m×V0×1000000
ʽ��:
Xi——�����������������ĺ�������λΪ��ÿ�ٿ�(g/100g);
Ci——�ɱ����߲�òⶨ��Һ����������������������λΪ��(μg);
V——���ⶨ��Һ�Ķ����������λΪ����( mL);
F——���ⶨ��Һ��ϡ�ͱ���;
V0——��Ʒ���ⶨʱ��ȡ���������λΪ����( mL);
m——�����ij�����������λΪ��( g);
100——��λת��;
1000000——��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
�ڶ��� ��ЧҺ��ɫ��-�����ⷨ
8 ԭ��
�����е��������������ܽ⡢ϡ�͡����˺�ʹ�þ������������ĸ�ЧҺ��ɫ���Ǽ�⣬����ɫ��ı���ʱ�䶨�ԣ���귨������
9 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
9.1 �Լ�
9.1.1���������(����≥99.5%)��
9.1.2���棨ɫ�״�����
9.1.3ʯ���ѣ��г� 30°C~60°C��
9.2 �Լ�����
�����������Һ����ȡ3.48g���������(����≥99.5%)��2000mLˮ�ܽ⣬Ũ��Ϊ10mmol/L��
9.3 ��Ʒ
D-���ᰱ��������������≥99.0%��
9.4 ����Һ������
9.4.1 ���ᰱ��������������Һ(1.0 mg/mL)����ȷ����105�����4Сʱ�����ص����ᰱ�������Ƕ���Ʒ10mg������10mL����ƿ�У���ˮ�������̶ȣ�ҡ�ȣ����á�
9.4.2���ᰱ������������Һ����ȡ���ᰱ��������������Һ0.5��1.0��1.5��2.0��4.0mL����10mL��ƿ�У��������ඨ����10ml����Ϊ����Һ��
10 �����豸
10.1 ��ЧҺ��ɫ���ǣ�������������DAD�������
10.2 ��ƽ������Ϊ0.1 mg��
10.3 �����䡣
10.4 ��������ϴ����
10.5 ���Ļ���ת�� ≥3000 r/min��
11 ��������
11.1 �����Ʊ�
11.1.1 ��������
��5.1.1��������ȡ����Һ��������50mL��ƿ�У���������ϡ�����̶Ⱥ�ҡ������0.45μm��Ĥ���˺����ù���Ʒ��Һ��
11.2 ɫ�ײο�����
11.2.1 ɫ������Cl8 4.6mm×250mm���������
11.2.2 ���£�35�档
11.2.3 ��Ⲩ����192nm��
11.2.4 �����ࣺ����+�����������Һ=10+90��
11.2.5 ���٣�0.8mL/min��
11.2.6 ��������10μL��
11.3 ������������
��10 μL�ı�ϵ�и�Ũ����Һ(9.4.2)��ע��Һ��ɫ�����У������Ӧ�ķ�������Ա���ҺŨ��Ϊ�����꣬�Է����Ϊ�����꣬���Ʊ����� (����ҺҺ��ɫ��ͼ����¼ A ��ͼ A.1 )��
11.4 ������Һ�IJⶨ
��10 μL����������Һ(11.1.1)ע��Һ��ɫ�����У��Ա���ʱ�䶨�ԣ���÷���������ݱ����ߵ�������Һ���ᰱ����������Ũ��(��Ʒ��ҺҺ��ɫ��ͼ����¼A��ͼA.2)��
12 ��������ı���
���������������ǣ������ᰱ�������Ǽƣ�������ʽ(2)����:
Ci ×V×F×100
Xi = ..........................................(2)
m×1000000
ʽ��:
Xi——�����������������ĺ�������λΪ��ÿ�ٿ�(g/100g);
Ci——�ɱ����߲�òⶨ��Һ�����ᰱ������������������λΪ��(μg/mL);
V——���ⶨ��Һ�Ķ����������λΪ����( mL);
F——���ⶨ��Һ��ϡ�ͱ���;
m——�����ij�����������λΪ��( g);
100——��λת��;
1000000——��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
13 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
��¼ A
����Һ��������Һ����Һ��ɫ��ͼ
A.1 ���ᰱ������������Һɫ��ͼ����ͼ A.1��

ͼ A.1 ���ᰱ������������Һɫ��ͼ
A.2 ����������������������Һɫ��ͼ����ͼ A.2��

ͼ A.2 ����������������������Һɫ��ͼ
��ʮ��������ʳƷ�������ƵIJⶨ
Determination of total triterpenes in health food
1 ��Χ
�������涨�˱���ʳƷ�������Ƶ�����-�ɼ��ֹ��Ȳⶨ������
��������������������ɷ�Ϊ��Ҫԭ�ϵı���ʳƷ�������ƺ����IJⶨ��
2 ԭ��
�������ȷ���ȡ�������������ʣ��ڸ�����������������ȩ��Ӧ������ɫ���ʡ����ܹ���Ϊ����Ʒ�����÷ֹ��ȷ��ⶨ��������548nm�����µ�����Ƚ��ж�����
3 �Լ��Ͳ���
��������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
�Լ�
3.1.1 ��������( CH3COOCH2CH3)��
3.1.2 �ȷ�(CHCl3)��
3.1.3 ������(CH3COOH)��
3.1.4 �����ᣨHClO4��
3.1.5 ���ȩ��C8H8O3��
�Լ�����
5%���ȩ��������Һ������Һ����ǰ���ơ����ܳ�ȡ���ȩ 0.5g���ӱ�����ʹ�ܽ�� 10mL�����á�
��Ʒ
�ܹ��ᣨC30H48O3�����й�ʳƷҩƷ�춨�о�Ժ��������≥93.8%��
����Һ������
�ܹ������Ʒ��Һ(100μg/mL)�����ܳ�ȡ�ܹ������Ʒ10mg����100mL����ƿ�У������������ܽⲢϡ�����̶ȣ�ҡ�ȡ�
4 �����豸
4.1 ����-�ɼ��ֹ��ȼơ�
4.2 ������ƽ����ȷ��0.1 mg��
4.3 ����ˮԡ����
4.4 ��������ϴ��
4.5 ���Ļ�
5 ��������
5.1�����Ʊ�
5.1.1 ��������
ȡһ�����������������ܳƶ��������������Ƶ���ԼΪ0.5��5mg������50mL��ƿ�У����ȷ�Լ30mL����������30 min�����䣬���ȷ����̶ȣ�ҡ�ȡ�
5.1.2 ������Ʒ
ȷ��ȡ�������������������������Ƶ���ԼΪ0.5��5mg��������100 mL����ƿ�У������������ܽⲢϡ�����̶ȣ�ҡ�ȡ�
5.2 �����ߵ�����
�ֱ�����ȡ�ܹ������Ʒ��Һ0.1��0.2��0.4��0.8��1.0mL���������У���60��ˮԡ�����ɣ����ܼ���0.4mL5%���ȩ��������Һ��ת��������ʹ�����ܽ⣬�پ��ܼ�1.0mL�����ᣬ���Ⱥ�����10mL������ɫ���У���60��ˮԡ����15min��ȡ������ԡ��ȴ���ܼ��������5.0mLҡ�ȣ�15min����1cm��ɫ����548nm�����ⶨ�����, �������Ϊ�����ꡢŨ��Ϊ��������Ʊ����ߡ�
5.3 ������Һ�IJⶨ
������ȡ5.1������������Һ1.0mL���������У���60��ˮԡ�����ɡ���5.2�����ߵ��������£���“���ܼ���0.4mL5%���ȩ��������Һ……”��ͬ���������ⶨ����ȣ��ӱ������϶�������Ʒ��Һ���ܹ���ĺ�����
6 ��������ı���
�����������ƺ�������ʽ����:
Xi= 
Xi-�����������ƺ��������ܹ���ƣ�����λΪ��ÿ�ٿˣ�g/100g����
Ci-�ɱ����߲�òⶨ��Һ����������������λΪ��(μg)��
m-�����ij�����������λΪ��(g)��
V1-�����������������λΪ������mL����
V2-�ⶨ�������������λΪ������mL����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
8 ����
������������Ϊ1.5μg
��ʮ�ġ�����ʳƷ��������IJⶨ
Determination of Content of Cordycepin in health food
1 ��Χ
�������涨�˱���ʳƷ���������Һ��ɫ�ײⶨ������
�����������ڱ���ƷʳƷ��������IJⶨ��
2 ԭ��
��������ˮ�ܽ⣬��ƫ������Һ�����������ʣ�����ɫ���룬���Ʒ�ı���ʱ��Ƚ϶��ԣ��Է������귨������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1 ƫ���ᣨHPO3������������
3.1.2 �״�(CH3OH)��ɫ�״���
3.2 �Լ�����
ƫ������Һ(30.0g/L)����ȡ30.0gƫ��������װ��Լ600mLˮ������ƿ�У��ڴ����������������ܽ⣬ת����1000mL����ƿ�У���ˮ������1000mL�����ȡ�
3.3 ��Ʒ
�����(C10H13N5O3)������≥98.0%��
3.4 ����Һ������
3.4.1����ر�������Һ(0.5 mg/mL)����ȡ��Ʒ25mg��50��������ƿ�У���ˮ�ܽⲢ�������̶� ��ҡ�ȡ�����ҺӦ������-18°C �����С�
3.4.2 �����������Һ����ȡ����ر�������Һ0.10mL��0.20mL��0.5mL��1.0mL��2.0mL��5.0 mL �� 50.0 mL ����ƿ�У���ˮ���ݣ���Ũ��Ϊ 1 mg/mL��2mg/mL��5mg/mL��10 mg/mL��20 mg/mL��50 mg/mL �ı�����Һ������ʱ���ơ�
4 �����豸
4.1 ��ЧҺ��ɫ���ǣ� ������������UV����
4.2 ��ƽ:����Ϊ 1 mg �� 0.1 mg��
4.3 ��������ϴ����
4.4 ���Ļ�:ת�� ≥4000 r/min��
4.5 ������������
5 ��������
5.1 �����Ʊ�
5.1.1 ��������
5.1.1.1 ������Ʒ��ȡ20������Ƭ�������������з��顢���ȣ���ȡ���̬�������ȣ������ҳ�ȡ�������ȷ��ȡ��������1g���ɸ�����Ʒ�к�����������ȷ��0.001g������50mL����ƿ�У���ˮ30mL�����ȣ�������30min������30.0g/Lƫ������Һ��3.2��1.0mL����������ˮϡ�����̶ȣ�ҡ�ȡ���Ʒ��0.45μ�� ��Ĥ���˺��Һ��ɫ������
5.1.1.2 Һ����Ʒ��ȷ��ȡһ����ҡ�Ⱥ������10 mL���ɸ�������������������50 mL����ƿ�У���ˮ20mL�����ȣ�������30min������30.0g/Lƫ������Һ��3.2��1.0mL����������ˮϡ�����̶ȣ�ҡ�ȡ���Ʒ��0.45μ�� ��Ĥ���˺��Һ��ɫ������
5.2ɫ�ײο�����
5.2.1 ɫ������C18����4.6×250 mm, 5 μm��ͬ������ɫ������
5.2.2 ���£�30 ��
5.2.3 ��Ⲩ����260 nm
5.2.4 �����ࣺ�״�-ˮ=15:85
5.2.5 ���٣�1.0 mL/min
5.2.6 ��������10 mL
5.3 �����ߵ�����
��10 μL�ı�ϵ�и�Ũ����Һ(3.4.2) ��ע��Һ��ɫ�����У������Ӧ�ķ�������ߣ� �Ա�����Һ��Ũ��Ϊ�����꣬�Է��������Ϊ�����꣬���Ʊ����� (����ҺҺ��ɫ��ͼ����¼ A ��ͼ A.1 )��
5.4 ������Һ�IJⶨ
�� 10 μL ����������Һ(5.1.1)ע��Һ��ɫ�����У��Ա���ʱ�䶨�ԣ���÷�������ߣ����ݱ����ߵõ�����Һ�������Ũ��(��Ʒ��ҺҺ��ɫ��ͼ����¼A��ͼA.2)��
6 ��������ı���
�����������������ʽ(1)����:
Ci ×V×100
Xi = ..........................................(1)
m×1000000
ʽ��:
Xi——������������ĺ�������λΪ��ÿ�ٿ�����ÿ������(g/100g/g/100mL);
Ci——�ɱ����߲�òⶨ��Һ���������Ũ�ȣ���λΪ��ÿ����(mg/mL);
V——���ⶨ��Һ�����ն����������λΪ����( mL);
m——�ⶨ��������������λΪ�������( g��mL);
100——��λת��;
1000000——��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
8 ����
��������Ϊ1 g���������Ϊ 50 mLʱ�������������Ϊ0.0010 g/100g��
��¼ A
����Һ��������Һ����Һ��ɫ��ͼ
A.1 ���������Һɫ��ͼ����ͼ A.1�� 
ͼ A.1 ���������Һɫ��ͼ
A.2 �����������������Һɫ��ͼ����ͼ A.2��

ͼ A.2 �����������������Һɫ��ͼ
��ʮ�塢����ʳƷ�г����IJⶨ
Determination of mannitol in health food
1 ��Χ
�������涨�˱���ʳƷ�г���ᣨ��¶�����ĸ�ЧҺ��ɫ���ⶨ������
�����������ڱ���ʳƷ�г���ᣨ��¶���������IJⶨ��
2 ԭ��
�����еij���ᾭ��ȡ���ڰ���ɫ�����Ϸ��룬��������ɢ��������⣬���ݱ���ʱ�䶨�ԣ������߷�������⡣
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.1 �Լ�
3.1.1 ���棨C2H3N����ɫ�״���
3.1.2 ��ˮ�Ҵ�(C2H5OH)��
3.2 �Լ�����
��ȡҺ��ȡ40mLˮ��60mL��ˮ�Ҵ���3.1.2����ϣ����á�
��Ʒ
����ᣨD-��¶��������Ʒ������≥99%��
����Һ������
��������Һ��10.0mg/mL���� ȷ����������Ʒ0.250 g������ˮ�ܽ����ˮ������25 mL�����ó�������Һ
��ϵ����Һ�Ʊ���������ȡ��������Һ0.1mL��0.2mL��0.5mL��1.0mL��2.0mL��4.0mL�ֱ�����10mL����ƿ�У���ˮ�������̶ȣ�ҡ�ȡ�����ʱ���ơ�
4 ����
4.1 ��ЧҺ��ɫ���ǣ� ��������ɢ��������ELSD����
4.2 ��������ϴ����
4.3 ���Ļ���ת��≥4000r/min��
4.4 ������ƽ������0.01 mg��0.1 mg
5 ��������
5.1 ��������
5.1.1 ������Ʒ��ȡ20������Ƭ�������������з��顢���ȣ���ȡ���̬�������ȣ������ҳ�ȡ�������ȷ��ȡ��������0.5 g���ɸ�����Ʒ�к�����������ȷ��0.001g������Բ����ƿ�У�����25mL��ȡҺ��3.2�������ȣ�������ˮԡ����60min��ȡ������ȴ������������ʧ��������ҡ�ȣ���0.45μm����Ĥ���ˣ���Һ��������
5.1.2 Һ����Ʒ��ȷ��ȡһ����ҡ�Ⱥ������10 mL���ɸ�������������������25 mL����ƿ�У�������ˮ�Ҵ���3.1.2��10mL�����ȣ�������30min��ȡ������ȴ���������̶ȡ����Ⱥ�0.45 μm��Ĥ���ˣ���Һ��ɫ�����á�
5.2 ɫ�ײο�����
5.2.1 ɫ����������Һ��ɫ������4.6×250 mm, 5 μm��ͬ������ɫ������
5.2.2 �����ࣺ������AΪˮ��������BΪ���档ˮ-����=25:75��
5.2.3 ���£�30�档
5.2.4 ���٣�1.0 mL/min��
5.2.5 ��������10 mL��
5.2.6 ������ɢ���������������٣�1.60slm��Ư�ƹ��¶ȣ�60�棬 ���棺1 ��
5.3 �������Ʊ�
�ֱ�����һϵ�г�������Һ���ڸ��������������½���Һ��ɫ�������Գ�����Ũ�ȣ�C���Ķ���LogCΪ�����꣬��Ӧ��ɫ�������A���Ķ���LogAΪ�����꣬���Ʊ����ߡ�
6 �ⶨ
ȡ10 μL����Һ��������Һע��ɫ�����У��Ա���ʱ�䶨�ԣ���������������ϵ�бȽ϶�����
7 �������
�����г���Ậ����ʽ(1)����:
Xi = ..........................................(1)
..........................................(1)
ʽ��:
Xi——�����г����ĺ�������λΪ��ÿ�ٿ˻��ÿ�ٺ���(g/100g��g/100mL);
Ci——�ɱ����߲�òⶨ��Һ�г�����Ũ�ȣ���λΪ����ÿ����( mg/mL);
V——���ⶨ��Һ�Ķ����������λΪ����( mL);
m——�����ij�����������λΪ�˻����( g��mL);
100——��λת��;
1000——��λת����
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
8 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 10% ��
9 ����
�����������Ϊ0.1μg��
������ɢ����������Ӧֵ��y���뱻����Ũ�ȣ�x���Ĺ�ϵ������������Ʒ�ƺͽṹ�IJ�ͬ����������ָ����ϵ����y=axb��Ҳ�ɸ���ʵ�����ѡ����ʵĹ�ϵ���ߡ�
��¼ A
����Һ��������Һ����ɫ��ͼ
A.1 ���������Һɫ��ͼ����ͼ A.1��

ͼA.1 ��������Һɫ��ͼ
A.2 ���г�����������Һɫ��ͼ����ͼ A.2��

ͼA.2 ���г�����������Һɫ��ͼ
��������
ʮһ���ܼ������IJⶨ
Determination of 11 kinds of
Residual Organic Solvents
1 ��Χ
�����涨�˱���ʳƷ���״������������춡�����ȷ¡������顢�ױ����Զ��ױ����ڶ��ױ�������ϩ��1,2-���һ����Ͷ���ϩ��11���ܼ�����������ɫ�ײⶨ������
���������ڱ���ʳƷ���״������������춡�����ȷ¡������顢�ױ����Զ��ױ����ڶ��ױ�������ϩ��1,2-���һ����Ͷ���ϩ��11���ܼ������IJⶨ��
2 ԭ��
��Ʒ��50% N,N-������������Һ��ȡ�������ö���-����ɫ���ⶨ������귨������
3 �Լ��Ͳ���
ע����������˵���������������Լ���Ϊ��������ˮΪ GB/T6682 �涨��һ��ˮ��
3.5 �Լ�
N,N-����������(HCON(CH3)2)��ɫ�״�
3.6 �Լ�����
50%��N,N-������������Һ�� 500 mL N,N-������������500 mLˮ��ֻ��ܻ�ϡ�
3.7 ��Ʒ
�״���CH4O������������CH3(CH2)3OH�����춡����CH3CH(CH3)CH2OH�����ȷ£�CHCl3���������飨C6H14�����ױ���C7H8�����Զ��ױ���C8H10�����ڶ��ױ���C8H10��������ϩ��C8H8����1,2-���һ�����C10H14���Ͷ���ϩ����C10H10����Ʒ������≥97.0%��
3.8 ����Һ������
3.4.1 ������Һ���ֱ�ȷ��ȡ2500 mg(��ȷ��0.1 mg) �״���1200 mg(��ȷ��0.1 mg)���������춡�����ȷ¡�120 mg(��ȷ��0.1 mg)������Ͷ���ϩ����100 mg(��ȷ��0.1 mg)�ױ����Զ��ױ����ڶ��ױ�������ϩ��1,2-���һ�����100 mL����ƿ�У���N,N-�����������������̶� ��ҡ�ȡ����״�Ũ��Ϊ25 mg/mL�����������춡�����ȷ�Ũ��Ϊ12 mg/mL��������Ͷ���ϩ��Ũ��Ϊ1.2 mg/mL���ױ����Զ��ױ����ڶ��ױ�������ϩ��1,2-���һ���Ũ��Ϊ1mg/mL������Һ��4°C������
3.4.2������м�Һ���ֱ�ȷ��ȡ2.0 mL�״�������Һ��1.0 mL ���������춡�����ȷ¡������顢����ϩ�����ױ����Զ��ױ����ڶ��ױ�������ϩ��1,2-���һ�����������Һ��ͬһ100 mL ����ƿ�У���50%��N,N-������������Һ�������̶� ��ҡ�ȡ��û�ϱ��м�Һ���״�Ũ��Ϊ500 μg/mL�����������춡�����ȷ�Ũ��Ϊ120 μg/mL�������顢����ϩ��Ũ��Ϊ12.0 μg/mL���ױ����Զ��ױ����ڶ��ױ�������ϩ��1,2-���һ���Ũ��Ϊ10.0 μg/mL�����м�Һ����4°C������
3.4.3 ��ϱ�����Һ��ȷ��ȡ��ϱ��м�Һ0.05 mL��0.20 mL��0.50 mL��1.0 mL��2.0 mL��5.0 mL��10.0 mL ����ƿ�У���50% N,N-�������������ݣ����״�Ũ��Ϊ2.50 μg /mL ��10.0 μg /mL��25.0 μg /mL��50.0 μg /mL��100 μg /mL��250 μg /mL�����������춡�����ȷ�Ũ��Ϊ0.60 μg /mL ��2.40 μg /mL��6.00 μg /mL��12.0 μg /mL��24.0 μg /mL��60.0 μg /mL�������顢����ϩ��Ũ��Ϊ0.06 μg /mL ��0.24μg /mL��0.60 μg /mL��1.20 μg /mL��2.40 μg /mL��6.00 μg /mL���ױ����Զ��ױ����ڶ��ױ�������ϩ��1,2-���һ���Ũ��Ϊ0.05 μg /mL ��0.20 μg /mL��0.50 μg /mL��1.00 μg /mL��2.00 μg /mL��5.00 μg /mL�ı�����Һ������ʱ���ơ�
4 �����豸
4.1 ����ɫ������������������ӻ������( FID)��
4.2 �����Զ���������
4.3 ����ƿ��20 mL���䱸���ǺͲ��������ܼ������Ķ��������֬��������
4.4 ��ƽ������Ϊ 1 mg �� 0.1 mg��
4.5 ���Ļ���ת�� ≥4000 r/min��
4.6 ��������ϴ����
5 ��������
5.1 �����Ʊ�
5.1.1 �������
��ȡ��Ʒ0.5 g (��ȷ��0.001 g) ��20 mL����ƿ�У�����5 mL50% N,N-������������Һ��ҡ�Ⱥ��ܷ�����������10 min�����á�
5.1.2 Һ�����
��ȡ��Ʒ1.0 g��2.0 g (��ȷ��0.001 g) ��10 mL����ƿ�У�����50% N,N-������������Һ�������̶���ҡ�ȣ�ȷ��ȡ5 mL��Һ��20mL����ƿ�У��ܷ⣬���á�
5.2���������
5.2.1���ս����ο�����
a) ƽ��ʱ�䣺30 min��
b) ƽ���¶ȣ�90 °C��
c) ���������1.0 mL��
5.2.2ɫ�ײο�����
a) ɫ�������Լ���/�������Ҷ���Ϊ�̶����ëϸ����������Ϊ30 m���ھ�Ϊ0.32 mm��Ĥ���Ϊ0.50 μm�������൱����
b) �������¶ȣ���ʼ�¶�40 °C������5 min ��10 °C/min ������ 150 °C������1 min������20 °C/min����200 °C������2 min��
c) �������¶ȣ�200 °C��
d) �����ȣ�15��1
e) FID ������¶ȣ�250°C��
f) �������ߴ����������� 1.5 mL/min��β�� 30 mL/min��
g) ����������40 mL/min��
h) ����������300 mL/min��
5.3 �����ߵ�����
ȷ��ȡ3.4.2����Ũ�ȵı�������Һ5 mL���ڶ���ƿ�У���������ɫ�ײⶨ�������Ӧ�ķ������ �Ա�����Һ����Ӧ�ܼ���Ũ��Ϊ�����꣬�Է����Ϊ�����꣬���Ʊ����� (����Һ����ɫ��ͼ����¼ A ��ͼ A.1 )��
5.4 ������Һ�IJⶨ
��5.1����������Һ��������ɫ���Dzⶨ���Ա���ʱ�䶨�ԣ���÷���������ݱ������������и��ܼ������ĺ�����
6 ��������ı���
�����и��ܼ�����������ʽ(1)����:
ʽ����
X——�����и��ܼ������ĺ�������λΪ����ÿǧ��(mg/kg)��
C——�ɱ����߲�òⶨ��Һ�и��ܼ�������Ũ�ȣ���λΪ��ÿ����(μg/mL)��
m——�����ij�����������λΪ��( g)��
V——����ƿ�й�������Һ���������λΪ����(mL)��
���������ظ������»�õ����ζ����ⶨ���������ƽ��ֵ��ʾ��������λ��Ч���֡�
7 ���ܶ�
���ظ��������»�õ����ζ����ⶨ����ľ��Բ�ֵ����������ƽ��ֵ�� 15 % ��
8 ����
��������Ϊ0.5 g ʱ���״��ļ����Ϊ4 mg/kg��������Ϊ10 mg/kg�����������춡�����ȷ¼����Ϊ2 mg/kg��������Ϊ6 mg/kg�������顢����ϩ���ļ����Ϊ0.2 mg/kg��������Ϊ0.6 mg/kg���ױ����Զ��ױ����ڶ��ױ�������ϩ��1,2-���һ���������ϩ�������Ϊ0.08 mg/kg��������Ϊ0.30 mg/kg��
��¼ A
11���ܼ������ʵĵ�������ɫ��ͼ
11���ܼ��ı����ʵ�����ɫ��ͼ��ͼA.1

ͼA.1 11���ܼ������ʵ�����ɫ��ͼ
1�������� 2���״� 3���ȷ� 4���ױ� 5���춡�� 6���Զ��ױ� 7�������� 8���ڶ��ױ� 9������ϩ 10�����һ��� 11������ϩ��
��������
�˷ܼ���Υ���ɷֲⶨ
Determination of Stimulant and
Prohibited Components
�˷ܼ���Υ���ɷֲⶨ
|
��Ʒ����
|
�˷ܼ���Υ���ɷ�
|
���鷽�����
|
|
��������ƣ�͡�
��ǿ������
|
2-�DZ���ȥ���������� ; 2-���һ�ȥ���������� ; N-����ϩ���������� ; N-������������ ; N-ȥ�������Ƿ� ; N-ȥ�һ�-N-�������Ƿ� ; N-ȥ�һ������Ƿ� ; N-ȥ�һ�����Ƿ� ; N-�嶡���ʻ�-N-ȥ�һ�����Ƿ� ; N-����ȥ���������� ; N-�һ��������� ; O-ȥ�һ������Ƿ� ; �����Ƿ� ; �����Ƿ� ; ������������ ; ���������Ƿ� ; ����ऺ���Ƿ� ; �������Ƿ� ; ����N-ȥ�������Ƿ� ; �������Ƿ� ; �������������Ƿ� ; ����������������Ƿ� ; �������������Ī�����Ƿ� ; ������������ǻ���Ī�����Ƿ� ; ����������������Ƿ� ; ���������ǻ���Ī�����Ƿ� ; �������������Ƿ� ; ���������춡�������Ƿ� ; �ﲴ��͡ ; ��������Ƿ� ; �����ȥ�����͵��Ƿ� ; �����ȥ�һ����͵��Ƿ� ; �����Ƿ� ; �����Ƿ�N-������ ; �����ǷǶ����� ; �����Ƿ����ͪ ; �����Ƿ������������� ; ����Ƿ� ; ��Ī�����Ƿ� ; ����Ƿ� ; �����Ƿ� ; ���͵��Ƿ� ; ��������Ƿ� ; �����Ī�����Ƿ� ; ��������Ƿ� ; ����߷� ; ���Ƿ�̼���� ; �ȵ��Ƿ� ; �����Ƿ� ; �Ƿ������� ; �Ǻ���Ƿ� ; ��Ī�����Ƿ� ; ��Ī�����Ƿ� ; �����Ƿ� ; �ǻ������Ƿ� ; �ǻ���Ī�����Ƿ� ; �ǻ�����Ƿ� ; �ǻ���������Ƿ� ; �ǻ������Ī�����Ƿ� ; �ǻ��������Ƿ� ; �ǻ��ȵ��Ƿ� ; ����Ƿ� ; ȥ�����͵��Ƿ� ; ȥ����������Ƿ� ; ȥ����ົ������Ƿǻ��� ; ȥ���������� ; ȥ̼�����Ƿ� ; ȥ�һ����͵��Ƿ� ; ˫�ȵ��Ƿ� ; ˫ȥ̼�����Ƿ� ; ˫ͪ����Ƿ� ; �������� ; �������Ƕ��ȴ����� ; �������Ǽ��Ȼ��� ; ͪ����Ƿ� ; �����Ƿ� ; ����ົ���������Ƿ� ; α�����Ƿ� ; �ڵ��Ƿ� ; �����Ƿ� ; �����Ƿ�N-������ ; �����ǷǶ��������� ; �����Ƿ�����12 ; �����Ƿ�����14 ; �����Ƿ� ; �������Ƿ� ; ���������������� ; ���������Ƿ� ; �춡�������Ƿ� ; �����
|
ʳƷ���Ƿ������ʵIJⶨ(BJS 201805)��
����׳�����г�ҩ�������ǷǼ���������ļ�ⷽ��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2008016����
����׳�����г�ҩ��PDE5�����Ƽ��Ŀ��ټ�ⷽ��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2009030����
|
|
����
|
N,N-˫ȥ���������� ; N-��ȥ���������� ; �������� ; ������ͪ ; ����˾�� ; �������� ; �������� ; ��ɳ��� ; �л��������� ; �������� ; ��ŵ���� ; �������� ; �ҷ����� ; ��̪ ; ���� ; ����͡ ; ��Ī�������� ; ���������� ; ����Ƽ� ; ������ ; ��Ī�ǰ� ; �工��͡ ; �ȴ��������� ; �ȿ�ɫ�� ; ����� ; ��Ƽ� ; �շ���͡ ; ������� ; ȥ��α��Ƽ� ; α��Ƽ� ; �������� ; ������͡ ; �Ŵ�����
|
ʳƷ�����������Ȼ�����IJⶨ(BJS 201701)��
���Ʒ���֢���г�ҩ��������������Ƽ�ҷ������ļ�ⷽ��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2006004����
�����ౣ��ʳƷΥ������ҩ��ļ�ⷽ����ʳҩ�������2010��114�ţ���
�������г�ҩ��ʳƷ�з�̪����������������������ļ�ⷽ��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2012005����
|
|
������Ѫ�ǡ�
������Ѫ֬��
������Ѫѹ��
����˯��
|
�������� ; ������� ; ��˾���� ; ������ ; ���ȵ�ƽ ; ��ɳ���� ; �ͱ��� ; ���ͱ��� ; ����˫�� ; ������ͪ ; ���ȷ��� ; ������ ; ������ ; ����˫�� ; ������� ; �����ƽ ; ���б��� ; ������� ; ���в��� ; �����ͪ ; �������� ; �������� ; �ױ��Ƕ��� ; �������� ; ���ֶ� ; �������� ; ��Ѫƽ ; ����ͪ ; ��ͨ�� ; �工��͡�������� ; �ȱ����� ; �ȵ� ; ������ͪ ; �������� ; ������͡ ; ������� ; ��Ī��ƽ ; ��Ⱥ��ƽ ; ������ƽ ; ����� ; ���ټ� ; ������� ; ������ ; ɳ������ ; ˾�ɰͱ��� ; �ʺ��� ; ���ǻ��工��͡ ; �������� ; ������ƽ ; ������ ; ���� ; ����ͱ��� ; ������¡ ; ��ƥ��¡
|
����ʳƷ��75�ַǷ����ӻ�ѧҩ��ļ��(BJS 201710)��
�������г�ҩ�зǷ����ӻ�ѧҩƷ������鷽��(����ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2009029)��
�������г�ҩ�зǷ��������ᶡ���Ҳ�����鷽��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2011008����
�������г�ҩ������Ѫ���ౣ��ʳƷ�зǷ����Ӹ��в���IJ�����鷽��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2013001����
������Ѫ֬�ౣ��ʳƷΥ������ҩ��ļ�ⷽ����ʳҩ�������2010��114�ţ���
����ʳƷ�зǷ�����ɳ���������鷽����8����鷽����ʳҩ��ʳ����[2016]28�ţ���
��ѹ���г�ҩ�зǷ����ӻ�ѧҩƷ������鷽��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2009032����
��ѹ���г�ҩ������ѹ�ౣ��ʳƷ�зǷ��������ֶ�������ѧ�ɷּ�ⷽ��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2014008����
�������г�ҩ�зǷ����ӻ�ѧƷ��ⷽ��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2009024����
�������г�ҩ�ͱ���ʳƷ�зǷ������ʺ��ء���ƥ��¡���ȱ�������������¡�IJ�����鷽��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2012004����
����˯�����г�ҩ������Ʒ�зǷ�������ͨ�������ټ��������������鷽��������ʳƷҩƷ�ල������ҩƷ���鲹����鷽���ͼ�����Ŀ����2013002����
|
��������
��ͬ���ͼ��ض�ԭ������Ʒ��
����ѧ�����Ŀ
Hygienic Testing Items for Products with Different Dosage Forms and Specific RawMaterials
��ͬ���ͼ��ض�ԭ������Ʒ������ѧ�����Ŀ
|
|
��Ʒ����
|
�����Ŀ
|
|
1
|
����
|
ˮ�֡��ҷ�
|
|
2
|
�ڷ�Һ
|
�����Թ����pHֵ��������
|
|
3
|
Ƭ���ͽ��Ҽ�
|
����ʱ��
|
|
4
|
���
|
ˮ�֡��л���ũҩ���������������ε��飩
|
|
5
|
���
|
��ɢʱ��
|
|
6
|
��Ƭ
|
�ܻ���
|
|
7
|
������
|
�ܻ��ԡ�����
|
|
8
|
�Ƽ�
|
�ܹ��塢�ƾ��ȡ��״����軯��������
|
|
9
|
������������
|
��ۡ�������ֵ����Ѫ֬���Ʒ����̴���
|
|
10
|
��ҩ��
|
���������ε����Լ�ҩ��涨�������ũҩ���ؽ������к�Ԫ��
|
|
11
|
ƻ����ɽ�
|
չ��ù��
|
|
12
|
���Ʒ
|
����ù����M1��M2��������
|
|
13
|
���ۡ��佺���Ʒ
|
������
|
|
14
|
����Ʒ
|
�ӡ��顢��
|
|
15
|
����
|
����ù����B1��B2��G1��G2������ù��
|
|
16
|
ֲ����
|
����ù����B1����ۡ�������ֵ
|
��������
�ض�ԭ������Ʒ����Ч�ɷֺ�
��־���ɷּ����Ŀ
Testing Items for Functional and Iconic Components of Products with Specific RawMaterial
�ض�ԭ������Ʒ����Ч�ɷֺ���־���ɷּ����Ŀ
|
|
ԭ�����
|
�����Ŀ
|
|
1
|
Ӫ���ز����
|
��Ʒ�б�ʶ��Ӫ���أ�����ά���غͿ����ʣ�
|
|
2
|
��ӿƲ���
|
����
|
|
3
|
ަ�ࣨĢ���ȣ�
|
��ʳ��ά�����ǡ������ƣ���֥�ࣩ
|
|
4
|
�����
|
����
|
|
5
|
�쾰����
|
�쾰����
|
|
6
|
«����
|
«����
|
|
7
|
������
|
������
|
|
8
|
��������
|
�����ʡ����ܲ��ء�ά����B1��ά����B2
|
|
9
|
��Ҷ��
|
����
|
|
10
|
ħ����
|
��ʳ��ά
|
|
11
|
������
|
��ʳ��ά
|
|
12
|
��֬��
|
��ͪ������Ҵ�����������鲻�������֬ԭ�ϣ���
|
|
13
|
������
|
�工��͡
|
|
14
|
ֲ������
|
֬���ᡢά����E
|
|
15
|
��������
|
֬����
|
|
16
|
������
|
������
|
|
17
|
¹Ѫ��
|
�����ʡ�������
|
|
18
|
������
|
�̡�������
|
|
19
|
�����
|
ø������ø����������
|
|
20
|
�ߡ�Ы��
|
�����ʡ�������
|
|
21
|
����ϩ
|
����ϩ
|
|
22
|
��ʽ�
|
10-�ǻ���ϩ��
|
|
23
|
�仨�ۡ��佺
|
�ܻ�ͪ
|
|
24
|
���ʲ�Ʒ
|
�������ȣ���Ʒ��Ϊ����Ӧ���ԭ�ϵ���������
|
|
25
|
�����ʡ���������Ʒ
|
�����ʡ�������
|
|
26
|
�ʺ��ز�Ʒ
|
��Ʒԭ�ϣ��ʺ��أ����ṩԭ�ϴ���֤�������
|
|
27
|
��øQ10��Ʒ
|
��Ʒԭ�ϣ���øQ10�����ṩ�Ʊ����ռ�����֤�������
|
|
28
|
��������ɷֵ�ԭ��
|
������
|
|
29
|
������
|
��Ƥ������
|
��¼
��¼1 ���ֹ�Ч�ɷ�/��־�Գɷּ�ⷽ�����ұ�
|
���
|
��Ч�ɷ�/��־�Գɷ�
|
����
|
|
1
|
α-�����ᡢ��ʮ̼��ϩ�ᡢ��ʮ��̼��ϩ�ᡢ��ʮ��̼��ϩ��
|
GB 28404 |
|
2
|
ǰ������
|
GB/T 22244 |
|
3
|
���Ƥ�
|
GB/T 22245 |
|
4
|
�����
|
GB/T 22246 |
|
5
|
������
|
GB/T 22247 |
|
6
|
�ʲ���
|
GB/T 22248 |
|
7
|
���Ѻ���
|
GB/T 22249 |
|
8
|
��ԭ��
|
GB/T 22250 |
|
9
|
�����
|
GB/T 22251 |
|
10
|
��øQ10
|
GB/T 22252 |
|
11
|
�����ͪ
|
GB/T 23788 |
|
12
|
�ʺ���
|
GB/T 5009.170 |
|
13
|
���������绯ø(SOD)
|
GB/T 5009.171 |
|
14
|
���������ͪ
|
GB/T 5009.193 |
|
15
|
������IgG
|
GB/T 5009.194 |
|
16
|
��़����
|
GB/T 5009.195 |
|
17
|
����
|
GB/T 5009.196 |
|
18
|
�������ء��������ߴ������ᡢ��������������
|
GB/T 5009.197 |
|
19
|
����B12
|
GB/T 5009.217 |
|
20
|
����K1
|
GB 5009.158 |
|
21
|
����B6
|
GB 5009.154 |
|
22
|
����B2
|
GB 5009.85 |
|
23
|
����B1
|
GB 5009.84 |
|
24
|
����A��D��E
|
GB 5009.82 |
|
25
|
���ơ�þ�������ء��ơ��ѡ����������̡������ܡ�����ͭ��п���顢�����ȡ��⡢�ӡ������ࡢ���������衢Ǧ����
|
GB 5009.268 |
|
26
|
�;���
|
GB/T 22491 |
|
27
|
�;�����ѿ��
|
GB/T 20881 |
|
28
|
�;۹���
|
GB/T 23528 |
|
29
|
��֬�������֬���Ҵ�������֬������
|
GB 5009.272 |
|
30
|
���������غ����ᰱ��������
|
GB/T 20365 |
��¼2 ��������ָ���ⷽ��
| ��� | ����ָ�� | ���ü�ⷽ�� | ��� | ����ָ�� | ���ü�ⷽ�� |
| 1 | Ǧ | GB 5009.12 | 11 | ˮ�� | GB 5009.3 |
| 2 | �� | GB 5009.11 | 12 | �ҷ� | GB 5009.4 |
| 3 | �� | GB 5009.17 | 13 | ������ֵ | GB 5009.227 |
| 4 | �� | GB 5009.15 | 14 | ��� | GB 5009.229 |
| 5 | �� |
GB 5009.123
���л�����ҩ�䡷
|
15 | ����ʱ�� | ���л�����ҩ�䡷 |
| 6 | ������ | GB 5009.19 | 16 | ��ɢʱ�� | ���л�����ҩ�䡷 |
| 7 | �ε��� | GB 5009.19 | 17 | pHֵ | GB 5009.237 |
| 8 | ����ù�� | GB 5009.222 | 18 | �ܻ��� | ���л�����ҩ�䡷 |
| 9 | ����ù����M1��M2 | GB 5009.24 | 19 | ���� | ���л�����ҩ�䡷 |
| 10 | ����ù����B1 | GB 5009.22 | 20 | չ��ù�� | GB 5009.185 |

������ ��